Silencing phospholipid scramblase 1 expression by RNA interference in colorectal cancer and metastatic liver cancer
2012-07-07WeiCuiShiYongLiJunFengDuZiManZhuandPingAn
Wei Cui, Shi-Yong Li, Jun-Feng Du, Zi-Man Zhu and Ping An
Beijing, China
Silencing phospholipid scramblase 1 expression by RNA interference in colorectal cancer and metastatic liver cancer
Wei Cui, Shi-Yong Li, Jun-Feng Du, Zi-Man Zhu and Ping An
Beijing, China
BACKGROUND:Phospholipid scramblase 1 (PLSCR1) not only participates in the transbilayer movement of phospholipids, but also plays a role in the pathogenesis and progression of cancers. The present study aimed to evaluate the effect of silencing PLSCR1 expression by RNA interference in colorectal cancer (CRC) and metastatic liver cancer.
METHODS:The expression of PLSCR1 in CRC and metastatic liver cancer samples was assessed by immunohistochemistry. The cultured cells with the highest expression were selected for subsequent experiments. We designed three siRNA oligonucleotide segments targeted at PLSCR1. Successful transfection was confirmed. The biological behavior of the cells in proliferation, adhesion, migration and invasion was determined.
RESULTS:PLSCR1 protein expression increased significantly in the majority of CRC and metastatic liver cancer samples compared with normal samples. Lovo cells had the highest expression of PLSCR1. The siRNA-390 oligonucleotide segment had the best silencing effect. After transfection, Lovo cell proliferation was significantly inhibited compared with the controls in the MTT assay. Laminin and fibronectin adhesion assays showed Lovo cell adhesion was also significantly inhibited. In the migration assay, the number of migrating cells in the PLSCR1 siRNA-390 group was 50±12, significantly lower than the number in the siRNA-N group (115±28) and in the control group (118±31). In an invasion test, the number of invading cells in the PLSCR1 siRNA-390 group was 60±18,significantly lower than that in the siRNA-N group (97±26) and the control group (103±24).
CONCLUSIONS:PLSCR1 is overexpressed in CRC and metastatic liver cancer. Silencing of PLSCR1 by siRNA inhibits the proliferation, adhesion, migration and invasion of Lovo cells, which suggests that PLSCR1 contributes to the tumorigenesis and tumor progression of CRC. PLSCR1 may be a potential gene therapy target for CRC and associated metastatic liver cancer.
(Hepatobiliary Pancreat Dis Int 2012;11:393-400)
phospholipid scramblase 1; colorectal cancer; metastatic liver cancer; RNA interference
Introduction
There are approximately 1 million new cases and 500 000 deaths annually from colorectal cancer (CRC). CRC is one of the most common malignant tumors. The incidence rate is gradually rising every year in developing countries, including China.[1]Today, CRC remains a serious health problem because the prognosis associated with this disease is poor, and about half of the newly-diagnosed patients die because of tumor recurrence and metastasis.[2,3]The initiation, development, local invasion, and distal metastasis of the tumor are controlled by multiple genes, and the expression of these genes is regulated by intrinsic and extrinsic factors.[4]Thus, clarification of the controlling factors and their patterns of expression may help in understanding the mechanism of carcinogenesis and metastasis of CRC, and thereby finding new targets for gene therapy in CRC patients.
Phospholipid scramblase 1 (PLSCR1) is a calciumbinding, multiple-palmitoylated type II endofacial plasma membrane protein. It participates in the transbilayer movement of phospholipids,[5]and its expression can be up-regulated by interferon, epidermal growth factor and leukemic cell differentiation-inducing agents.[6]PLSCR1interacts with several protein kinases including c-Abl, c-Src, protein kinase Cδ, and onzin, which contributes to cell signaling pathways.[7]Increasing evidence[7]also suggests that it plays an important role in cell proliferation, differentiation, and apoptosis, and also that it contributes to the pathogenesis and progression of cancers.
More recently, RNA interference has been implicated in the regulation of gene expression; RNA interference provides a simple, effective, specific, and important mechanism to silence gene expression.[8]RNA interference involves a nuclease to initially cut double-stranded RNA into small interfering RNA (siRNA) fragments with 21 to 25 nucleotides. Based on the mechanism of base pairing, the siRNA recognizes and cleaves the homogenous target mRNA molecule, which can silence the sequence-specific mRNA.[9]This mechanism is sequence-specific and causes downregulation of specific protein expression.[10,11]RNA interference technology is currently considered an important tool for functional genomic analyses and is also an important mechanism for specific gene-silencing therapeutics.[12-14]In the present study, the expression of PLSCR1 in CRC and metastatic liver cancer samples was investigated by immunohistochemistry. The siRNA technique was used to silence the expression of PLSCR1 in human CRC cells, and to evaluate the biological behavior of the cells in order to determine the role of PLSCR1 expression in tumor proliferation, adhesion, migration and invasion.
Methods
Tissue samples
Samples from 10 normal colon, 50 CRC, and 8 metastatic liver cancer patients (acquired from the Department of General Surgery in our hospital) were used in this analysis. All CRC patients, 27 women and 23 men, mean age 57 years (range 37-70), had histologically verified adenocarcinoma of the colon or rectum that was confirmed by pathologists. The 50 CRC specimens comprised 15 well-differentiated, 15 moderatelydifferentiated and 20 poorly-differentiated cancers. The specimens were fixed and then embedded in formalin and paraffin. All of the procedures were performed in accordance with theDeclaration of Helsinkiand were approved by the Ethics Review Board of our hospital.
Cell lines and cell culture
Colo-320, Sw620, HR8348, and Lovo cell lines (purchased from the type culture collection of the Chinese Academy of Sciences, Shanghai, China) were grown in DMEM with 10% fetal bovine serum (FBS) (Sigma, USA). All cells were cultured in a humidified incubator with 5% CO2at 37 ℃. Cells with mycoplasma and viral contamination were excluded throughout the experiments.
Immunocytochemistry
The Colo-320, Sw620, HR8348, and Lovo cells were fixed in 2% methanol for 20 minutes and washed three times with phosphate-buffered saline (PBS). After blocking with 1.5% goat serum for 60 minutes, the cells were incubated with anti-PLSCR1 antibody (Abcam, UK) at 4 ℃ overnight. The cells were then incubated with fluorescent, labeled secondary antibody (Zhongshan Golden Bridge Biotechnology, China) for 60 minutes at room temperature. All cells were observed under a fluorescence microscope and representative samples were selected and photographed. The tests were repeated three times.
The 3-µm thick tissue sections were cut from paraffin-embedded samples. The sections were first deparaffinized with xylene, and then rehydrated using an ethanol gradient. For antigen retrieval, the specimens were heated in a microwave oven (10 mmol/L citrate buffer, pH 6.0, 30 minutes, 600 W). Next, the sections were incubated with 3% H2O2at room temperature for 10 minutes to eliminate endogenous peroxidase activity. The samples were then incubated with primary anti-PLSCR1 antibody (Abcam, UK) overnight at 4 ℃. Finally, biotinylated secondary antibody (Zhongshan Golden Bridge Biotechnology, China) was added at room temperature and incubated for 30 minutes, and then developed by 3, 3'-diaminobenzidine (DAB) for 5 minutes, washed in running water, and counterstained with hematoxylin. Representative samples were selected and photographed. The evaluation standards were from Kuo's article.[15]
RNA interference
The PLSCR1 siRNAs were produced by Genepharm (Shanghai, China) and used forin vitrogene transfection. The cells were transfected with siRNA using the oligofectamine transfection reagent (Genepharm). The three siRNAs used were as follows: siRNA-390, sense 5'-UGG ACA AAC AAA ACU CAC ATT-3', anti-sense 5'-UGU GAG UUU UGU UUG UCC ATT-3'; siRNA-851, sense 5'-GGG CCA UCU AGA CCU UUU ATT-3', antisense 5'-UAA AAG GUC UAG AUG GCC CTT-3'; and siRNA-911, sense 5'-GGA GAG ACC ACU AAG AUG UTT-3', anti-sense 5'-ACA UCU UAG UGG UCU CUC CTT-3'. The sequence of siRNA-N was as follows: sense 5'-UUC UCC GAA CGU GUC ACG UTT-3', anti-sense 5'-ACG UGA CAC GUU CGG AGA ATT-3'. Before transfection, 5×104cancer cells per well were seededin six-well plates and incubated overnight until they reached 70% confluence. Then, they were transfected by following the manufacturer's instructions. Real-time PCR and Western blotting analyses were used to assess the success of transfection.
Real-time PCR analysis
Before PCR amplification, total RNA was prepared using Trizol reagent (Invitrogen, USA) and the cDNA was synthesized by reverse transcription using the PrimeScriptTMRT-PCR kit (TaKaRa, Japan), according to the manufacturer's instructions. cDNA was then amplified using a real-time PCR cycler (ABI, USA). The sequences of the PCR primers were: PLSCR1, 5'-cac cca tgt cta cca aag tt-3' and 3'-ctc tca aaa ttc cag tcc ag-5'. The reaction system was a mixture of 12.5 µL of 2×SYBR Green PCR Master mix, 100 nmol/L Primer A, 100 nmol/L Primer B, 1 µL cDNA, and dH2O up to 25 µL. The thermal profile was composed of 1 cycle at 95 ℃ for 5 minutes followed by 40 cycles at 94 ℃ for 20 seconds, 58 ℃ for 20 seconds, and 72 ℃ for 20 seconds, then 1 cycle at 72 ℃ for 5 minutes and at 55 ℃ for 10 seconds. The melting curve was from 65 ℃ to 95 ℃. The expression level of PLSCR1 was assessed by normalization of the cycle threshold (Ct) of the genes to that of the control gene (GAPDH).[16]∆Ct=CtPLSCR1-CtGAPDH, ∆∆Ct=∆Ctmin-∆Ctx, the relative quantification of the target=2-∆∆Ct.
Western blotting
Total protein was extracted after cell disruption, and its concentration was assessed. The proteins in the samples in each group were separated using 8% SDS-PAGE. The different proteins were transferred to PVDF membranes (Millipore, USA) and the membranes were then blocked. After blocking, a dilute solution of primary antibody (1:400 Abcam, UK) was incubated with the membrane under gentle agitation. The membrane was then exposed to HRP-conjugated secondary antibody (1:30 000 Zhongshan Golden Bridge Biotechnology, China), and the specific protein was checked with a SuperSignal protein detection kit (Zhongshan Golden Bridge Biotechnology). Then the membrane was stripped and re-probed by the same procedure using an antibody against GAPDH (1:1000).
MTT assays
Before the MTT assays, 1×104cancer cells per well were seeded into 96-well microtiter plates and incubated overnight. At 12, 24, 48, or 72 hours after siRNA transfection, 50 µL of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, Sigma, USA) (5 g/L) was added to each well for 4 hours. Then the formazan crystals were dissolved in 150 µL of dimethyl sulfoxide after aspirating the medium containing MTT. A spectrophotometer (Bio-Rad, USA) was used to determine the absorbance at 570 nm. All experiments were repeated three times, and the data were recorded as the mean of the three experiments.
Adhesion assays
A 96-well microtiter plate was coated with Matrigel (20 µg/well), laminin (Ln, 20 µg/well) or fibronectin (Fn, 10 µg/well). Then 6×104cancer cells per well plated onto these components. No chemicals for extracellular stimulation were added. The cells were permitted to adhere to each well for 2 hours at 37 ℃ and then gently washed three times in PBS. The adhesion of cancer cells to extracellular components was assessed by fixation with formalin and staining with hexamethylpararosaniline for 2 hours at 37 ℃. After 100 µL 2% SDS was added per well, a spectrophotometer (Bio-Rad, USA) was used to determine the absorbance at 570 nm. All experiments were repeated three times, and the data were recorded as the mean of the three experiments.
Transwell migration assays
Before seeding the harvested cancer cells, the transwells (Corning, USA) were pretreated with serumfree medium for 1 hour at 37 ℃, then 1×105cells in 2 mL of serum with free FBS albumin were added. Next, the transwells were inserted into 6-well plates with 1 mL conditioned medium containing 0.1% FBS and incubated for 24 hours at 37 ℃ with 5% CO2. The cells on the upper side of the filter were removed and those on the lower side were fixed in 95% ethanol and stained with hematoxylin and eosin (HE). The number of migrating cells was counted in 5 randomly-selected visual fields on the filter using an inverted microscope at 400× magnification. All experiments were repeated three times and the data were recorded as the mean of the three experiments.
Transwell invasion assays
After coating with matrigel (3.9 µg/µL, 60 µL to 80 µL, BD, USA) on the upper surface of a polycarbonate membrane, the transwell filters were incubated for 30 minutes at 37 ℃ until the gel solidified and acted as the extracellular matrix. Cancer cells, 5×104per mL of serum, were placed into the upper compartment with free FBS albumin, and 1 mL of conditioned medium with 0.1% FBS was placed in the lower compartment. The cells on the lower side of the filter were permitted to migrate at 37 ℃ with 5% CO2for 24 hours. The cells on the upper side of the membrane were eliminatedafter 24 hours. The cells on the lower side were fixed in 95% ethanol and stained with HE. The number of cells that had migrated was counted in 5 randomly-selected visual fields on the filter using an inverted microscope at 400× magnification. All experiments were repeated three times and the data were recorded as the mean of the three experiments.
Statistical analysis
All data were analyzed with SPSS 15.0 for Windows (SPSS Inc., Chicago, IL., USA). The Chi-square test was used to compare the PLSCR1 expression among various samples. An independentttest was performed to compare the differences between the groups. One-way ANOVA was used to analyze the significance in multiple groups. The LSD method of multiple comparisons was performed when the probability for ANOVA was statistically significant.Pvalue <0.05 was considered statistically significant.
Results
Expression of PLSCR1 in CRC and metastatic liver cancer
Immunohistochemical staining showed that the expression of PLSCR1 in CRC and metastatic liver cancer samples was significantly increased compared to normal mucosa, and that the PLSCR1 protein was mainly located at the cancer cell membrane (Fig. 1). The immunohistochemical staining intensity of PLSCR1 was absent or weak in most of the normal tissue samples (80.0%), positive in 23 of 50 (46.0%) in adjacent cancer tissues, positive in 43 of 50 (86.0%) in CRC samples, and positive in 7 of 8 (87.5%) in metastatic liver cancer samples. A significant difference in PLSCR1 expression level was found among the normal samples, adjacent cancer samples, CRC samples, and metastatic liver cancer samples (P<0.05). But the positive rate of PLSCR1 expression in well-differentiated, moderatelydifferentiated, and poorly-differentiated cancers was 80.0% (12/15), 86.7% (13/15), and 90.0% (18/20), respectively (P>0.05). No difference in PLSCR1 expression was found in cancers with varying differentiation.
Expression of PLSCR1 in CRC cells
All four CRC cell types showed PLSCR1 protein expression by immunocytochemistry and the protein was mainly located at the cell membranes as shown by immunohistochemistry. Lovo and HR8348 cells had more PLSCR1 protein expression than Colo-320 or Sw620 cells (Fig. 2). PCR and Western blotting also confirmed these results (Figs. 3 and 4). Lovo cells were chosen for the subsequent experiments because, of the four cell types investigated, they had the highest expression of PLSCR1 at both mRNA and protein levels.
Suppression of PLSCR1 expression by siRNAs
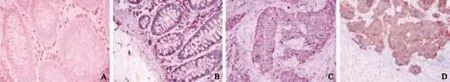
Fig. 1. PLSCR1 overexpressed in CRC and metastatic liver cancer. PLSCR1 immunohistochemical staining is presented for normal tissue (A), adjacent cancer tissue (B), CRC (C), and metastatic liver cancer (D). The localization of PLSCR1 was in the cell membrane (stained brown).
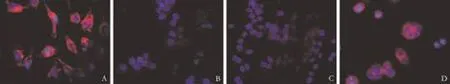
Fig. 2. Lovo (A) and HR8348 (D) cells demonstrated more expression of PLSCR1 protein than Colo-320 (B) and Sw620 (C) cells (red is positive).
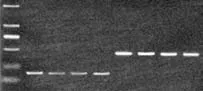
Fig. 3. Results of PCR analysis. 1-4: PLSCR1, 5-8: GAPDH; 1 and 5: Lovo; 2 and 6: Colo-320; 3 and 7: Sw620; 4 and 8: HR8348.

Fig. 4. Results of Western blotting analysis. 1: Lovo; 2: Colo-320; 3: Sw620; 4: HR8348.
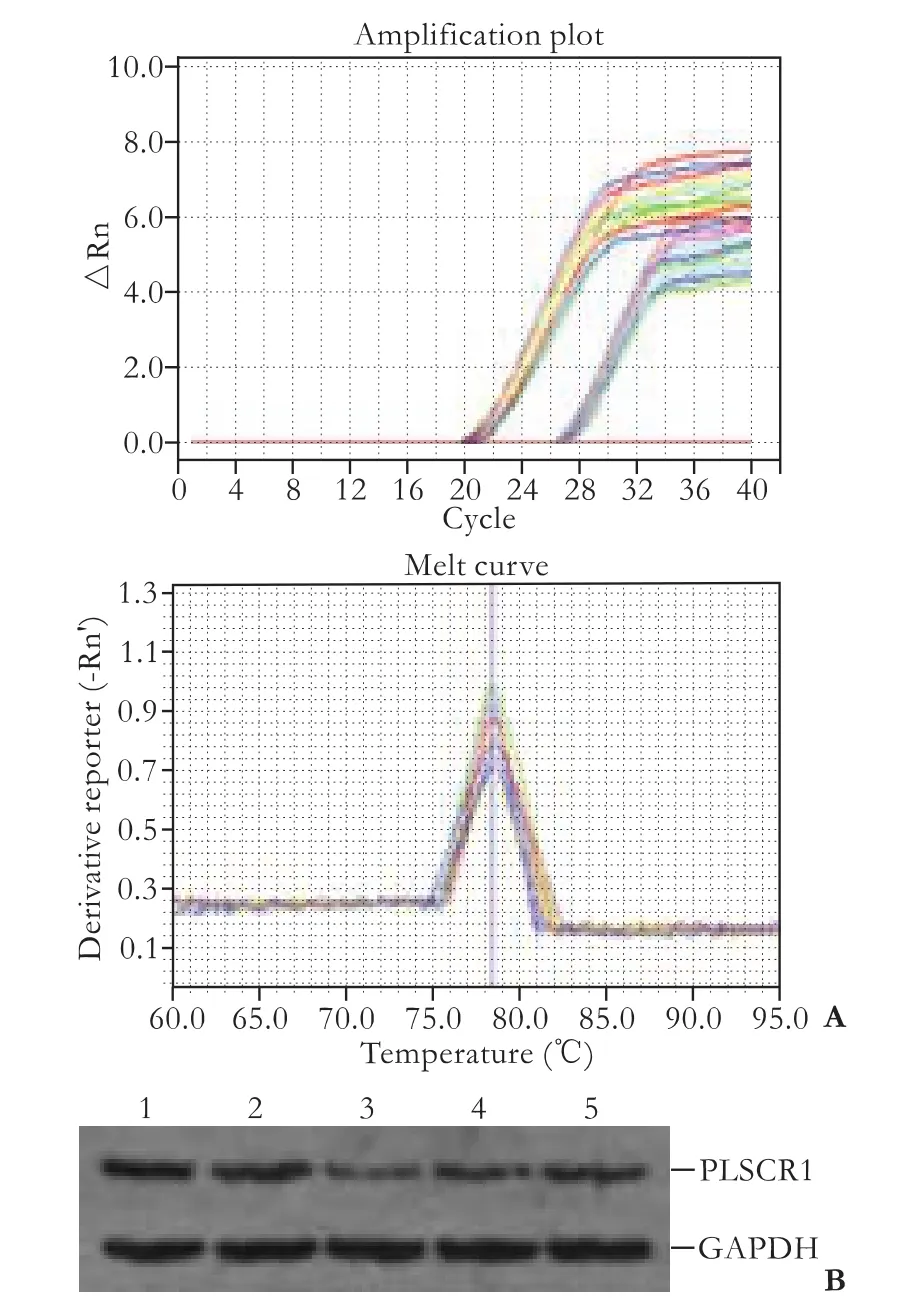
Fig. 5. Real-time PCR (A) and Western blotting analysis (B) of Lovo cells after transfection. 1: control; 2: siRNA-N; 3: siRNA-390; 4: siRNA-851; 5: siRNA-911.
Forty-eight hours after transfection, real-time PCR showed that the relative quantification of PLSCR1 RNA in controls was 4.4587±0.6585, in siRNA-N 3.9841± 0.0485, in PLSCR1 siRNA-390 0.5186±0.2327, in PLSCR1 siRNA-851 1.3224±0.2119, and in PLSCR1 siRNA-911 2.1770±0.3723. siRNA-390 inhibited the expression of PLSCR1 more significantly than siRNA-851 and siRNA-911 (P<0.05), while siRNA-N expression was not different from the controls, showing that siRNA-N did not affect PLSCR1 expression (Fig. 5A). These results, obtained using Western blotting, indicated that the PLSCR1 protein expression was silenced after siRNA-390 transfection. The expression of PLSCR1 was significantly down-regulated by 69.78% and 71.93% by siRNA-390 transfection, relative to the siRNA-N and control groups (Fig. 5B). siRNA-390 was therefore chosen for the subsequent experiments.
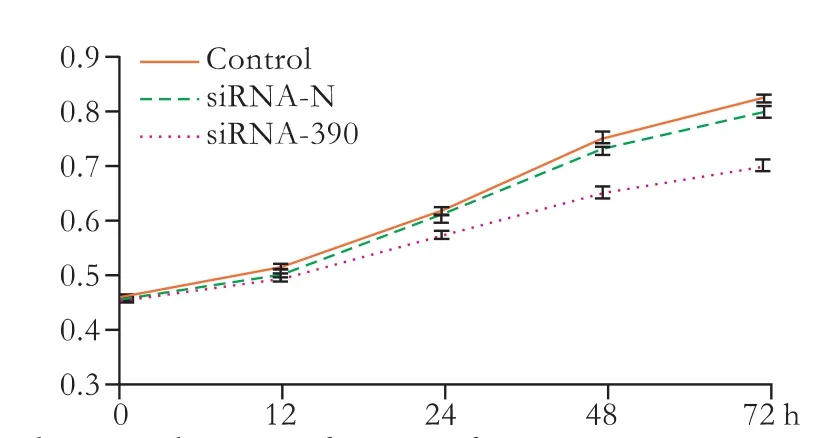
Fig. 6. The growth curve after transfection.
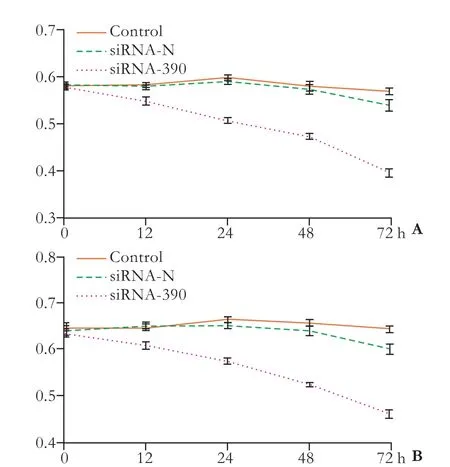
Fig. 7. Results of the Ln (A) and Fn (B) adhesion assays after transfection.
Effects of siRNA-390 on Lovo cell proliferation
The biological behavior of Lovo cells following siRNA-390 transfection was evaluated using cell proliferation assays. After transfection, Lovo cell proliferation was inhibited at 48 and 72 hours in the control group (P<0.05). In addition, no difference was found between the siRNA-N and control groups over the entire experimental period (Fig. 6). These effects were time-dependent, and showed that silencing PLSCR1 blocked Lovo cell proliferation.
Effects of siRNA-390 on Lovo cell adhesion
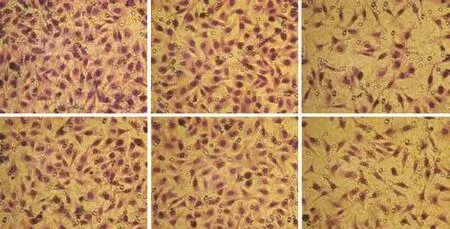
Fig. 8. The transwell assay showed that migration (A) and invasive (B) were was significantly inhibited in the siRNA-390 group compared with the control and siRNA-N groups.
Ln and Fn adhesion assays were performed to assess Lovo cell adhesion. After transfection, the assays showed that adhesion was inhibited at 48 and 72 hours in the control group (P<0.05). In addition, no difference was found between the siRNA-N and control groups throughout the entire experiment (Fig. 7). These effects were time-dependent, and the results showed that silencing PLSCR1 inhibited Lovo cell adhesion.
Effects of siRNA-390 on Lovo cell migration
After transfection, the transwell assay showed that migration was significantly inhibited compared with the control group. Representative micrographs of the transwell filters are presented in Fig. 8A. The number of migrating cells in the PLSCR1 siRNA-390 group was 50±12, markedly lower than that in the siRNA-N (115 ±28) and control groups (118±31) (P<0.05). There was no difference between cells in the siRNA-N and control groups.
Effects of siRNA-390 on Lovo cell invasion
Cell invasion was confirmed by the ability of cells to invade a matrix barrier; the barrier was composed of type IV collagens and laminins, which are the major components of the basement membrane. Representative micrographs of transwell filters are displayed in Fig. 8B. The number of invading cells in the PLSCR1 siRNA-390 group was 60±18, lower than that in the siRNA-N (97± 26) and control groups (103±24) (P<0.05). No difference was found between the siRNA-N and control groups.
Discussion
CRC is a serious health problem in all parts of the world.[1]Its treatment is appropriate for most patients, which mainly focuses on surgical removal of the entire tumor and adjacent lymph nodes, and radiation and chemotherapy. However, the prognosis in patients with CRC is still poor, mostly because of localized recurrence and liver metastasis. Thus, new therapeutic targets would be beneficial to CRC patients.
PLSCR1 is a calcium-binding, multiple-palmitoylated type II endofacial plasma membrane protein. Studies have revealed that it participates in the transbilayer movement of phospholipids, but more studies[6,7]propose that it takes part in cell proliferation, differentiation, apoptosis, and the pathogenesis and progression of cancers. Although PLSCR1 is found in different cells, its expression level may vary appreciably in different tissue types. A recent study[17]has shown that PLSCR1 may be a positive acute-phase protein. Its overexpression is associated with the differentiation of human myeloid leukemia cells into granulocytes,[18]and inhibits growth in ovarian carcinoma cells.[19]Moreover, the expression of PLSCR1 varies in different tumor diseases and inflammatory conditions. However, the function of PLSCR1 protein in the pathogenesis and progression of CRC is still unclear. The expression level of PLSCR1 is significantly elevated in malignant adenocarcinoma compared with normal colorectal mucosa, and its plasma level is increased in CRC patients, especiallyin those at an early stage.[15]Univariate analysis using the Cox regression model also indicated that increased PLSCR1 expression is associated with a poor prognosis,[15]suggesting that PLSCR1 overexpression may be an early but important marker in CRC. This also suggests that PLSCR1 overexpression may play a significant role in tumorigenesis and tumor progression.
In our study, PLSCR1 was overexpressed in CRC and metastatic liver cancer, but there was no correlation with the degree of cancer differentiation. Real-time PCR and Western blotting confirmed that PLSCR1 siRNA-guided RNA inhibition (RNAi) efficiently silenced PLSCR1 expression in Lovo cancer cells. To assess the changes in cell proliferation, adhesion, migration and invasion following RNAi, the appropriate assays were performed using Lovo cells. Our results showed that their proliferation, adhesion, migration and invasion were blocked after PLSCR1 siRNA transfection. This may be a result of PLSCR1 up-regulation of angiogenin, which enhances rRNA transcription, and thereby potentiates angiogenesis and tumor cell proliferation.[20]Another possible explanation for our results is that PLSCR1 could interact with activated epidermal growth factor receptors (EGFRs). Stimulation of cells expressing EGFRs on their surface by epidermal growth factor (EGF) causes an association of PLSCR1 with the activated EGFRs and Shc (an adaptor protein), which leads to tyrosine phosphorylation of PLSCR1.[21]Binding of EGF with EGFRs also causes the internalization of PLSCR1 from the plasma membrane in conjunction with EGFRs. While EGFRs are subsequently ubiquitinated and degraded, PLSCR1 is recycled from its endosomal compartment to the cell surface.[21]It was reported that activated Src kinase leads to the tyrosine phosphorylation of PLSCR1 found in EGF-treated cells and that this phosphorylation is needed for the interaction of PLSCR1 with Shc upon stimulation by EGF.[21]A third possible explanation for our results is that the human PLSCR1 gene is activated in response to cytokines such as interferons (IFNs). IFN-α, IFN-β and IFN-γ cause a substantial increase the expression of PLSCR1.[22]The high expression level of PLSCR1 when stimulated with IFNs in response to viral infection suggests that PLSCR1 is involved in the immune responses.[23]Suppression of PLSCR1 expression by RNAi may block the two signaling pathways and impair cellular immune responses, thus inhibiting Lovo cell proliferation, adhesion, migration and invasion.
In summary, PLSCR1 was associated with the pathogenesis and progression of CRC in ourin vitrostudy. Blocking PLSCR1 expression using the siRNA approach significantly reduced PLSCR1 protein levels and inhibited the proliferation, adhesion, migration and invasion of Lovo cells. Because Lovo cells are representative CRC cells, and mimic the biological behavior of CRC to some degree, PLSCR1 may be a potential therapeutic target for the treatment of CRC and metastatic liver cancer.
Contributors:LSY proposed the study. CW and LSY performed research and wrote the first draft. DJF, ZZM and AP collected and analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. LSY is the guarantor.
Funding:This work was supported by grants from the National Natural Science Foundation of China (81041025 and 81000189).
Ethical approval:This work was approved by the Ethics Review Board of the General Hospital of Beijing Military Command, Beijing, China.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005;55:74-108.
2 Wilke HJ, Van Cutsem E. Current treatments and future perspectives in colorectal and gastric cancer. Ann Oncol 2003;14:ii49-55.
3 Baca B, Beart RW Jr, Etzioni DA. Surveillance after colorectal cancer resection: a systematic review. Dis Colon Rectum 2011;54:1036-1048.
4 Matsubara N. Epigenetic regulation and colorectal cancer. Dis Colon Rectum 2012;55:96-104.
5 Sims PJ, Wiedmer T. Unraveling the mysteries of phospholipid scrambling. Thromb Haemost 2001;86:266-275.
6 Sahu SK, Gummadi SN, Manoj N, Aradhyam GK. Phospholipid scramblases: an overview. Arch Biochem Biophys 2007;462:103-114.
7 Huang Y, Zhao Q, Chen GQ. Phospholipid scramblase 1. Sheng Li Xue Bao 2006;58:501-510.
8 Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-811.
9 Martinez J, Patkaniowska A, Urlaub H, Lührmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 2002;110:563-574.
10 McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 2002;3:737-747.
11 Shi Y. Mammalian RNAi for the masses. Trends Genet 2003;19:9-12.
12 Grimm D, Kay MA. Therapeutic application of RNAi: is mRNA targeting finally ready for prime time? J Clin Invest 2007;117:3633-3641.
13 Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev 2007;59:75-86.
14 Zhao F, Zhang Q, Kang C, Cui X, Wang T, Xu P, et al. Suppression of matrix metalloproteinase-9 expression by RNA interference inhibits SGC7901 gastric adenocarcinomacell growth and invasionin vitroandin vivo. Med Oncol 2010;27:774-784.
15 Kuo YB, Chan CC, Chang CA, Fan CW, Hung RP, Hung YS, et al. Identification of phospholipid scramblase 1 as a biomarker and determination of its prognostic value for colorectal cancer. Mol Med 2011;17:41-47.
16 Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402-408.
17 Lu B, Sims PJ, Wiedmer T, Moser AH, Shigenaga JK, Grunfeld C, et al. Expression of the phospholipid scramblase (PLSCR) gene family during the acute phase response. Biochim Biophys Acta 2007;1771:1177-1185.
18 Nakamaki T, Okabe-Kado J, Yamamoto-Yamaguchi Y, Hino K, Tomoyasu S, Honma Y, et al. Role of MmTRA1b/ phospholipid scramblase1 gene expression in the induction of differentiation of human myeloid leukemia cells into granulocytes. Exp Hematol 2002;30:421-429.
19 Silverman RH, Halloum A, Zhou A, Dong B, Al-Zoghaibi F, Kushner D, et al. Suppression of ovarian carcinoma cell growth in vivo by the interferon-inducible plasma membrane protein, phospholipid scramblase 1. Cancer Res 2002;62:397-402.
20 Vinayagam A, Stelzl U, Foulle R, Plassmann S, Zenkner M, Timm J, et al. A directed protein interaction network for investigating intracellular signal transduction. Sci Signal 2011;4:rs8.
21 Nanjundan M, Sun J, Zhao J, Zhou Q, Sims PJ, Wiedmer T. Plasma membrane phospholipid scramblase 1 promotes EGF-dependent activation of c-Src through the epidermal growth factor receptor. J Biol Chem 2003;278:37413-37418.
22 Yokoyama A, Yamashita T, Shiozawa E, Nagasawa A, Okabe-Kado J, Nakamaki T, et al. MmTRA1b/phospholipid scramblase 1 gene expression is a new prognostic factor for acute myelogenous leukemia. Leuk Res 2004;28:149-157.
23 Dong B, Zhou Q, Zhao J, Zhou A, Harty RN, Bose S, et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J Virol 2004;78:8983-8993.
January 27, 2012
Accepted after revision May 11, 2012
Author Affiliations: Department of General Surgery, General Surgery Center of the PLA, General Hospital of Beijing Military Command, Beijing 100700, China (Cui W, Li SY, Du JF and An P); Department of Hepatobiliary Surgery, First Affiliated Hospital, General Hospital of the PLA, Beijing 100037, China (Zhu ZM)
Shi-Yong Li, Professor, Department of General Surgery, General Surgery Center of the PLA, General Hospital of Beijing Military Command, Beijing 100700, China (Tel/Fax: 86-10-66721188; Email: lsybz@126.com)
© 2012, Hepatobiliary Pancreat Dis Int. All rights reserved.
10.1016/S1499-3872(12)60197-0
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Gastric- and intestinal-type marker expression in invasive ductal adenocarcinoma of the pancreas
- Early changes of hepatic hemodynamics measured by functional CT perfusion in a rabbit model of liver tumor
- A common variant in the precursor miR-146a sequence does not predispose to cholangiocarcinoma in a large European cohort
- Muscarinic acetylcholine receptor M3 in proliferation and perineural invasion of cholangiocarcinoma cells
- Hepatocyte differentiation of mesenchymal stem cells
- Early control of short hepatic portal veins in isolated or combined hepatic caudate lobectomy