Ron receptor-dependent gene regulation in a mouse model of endotoxin-induced acute liver failure
2012-07-07RishikeshKulkarniLouisKutcherWilliamStuartDanielCarsonMikeLeonisandSusanWaltz
Rishikesh M Kulkarni, Louis W Kutcher, William D Stuart, Daniel J Carson, Mike A Leonis and Susan E Waltz
Cincinnati, USA
Ron receptor-dependent gene regulation in a mouse model of endotoxin-induced acute liver failure
Rishikesh M Kulkarni, Louis W Kutcher, William D Stuart, Daniel J Carson, Mike A Leonis and Susan E Waltz
Cincinnati, USA
BACKGROUND:Prior experimentation has shown that loss of the tyrosine kinase (TK) signaling domain of the Ron receptor leads to marked hepatocyte protection in a model of lipopolysaccharideinduced acute liver failure (ALF) in D-galactosamine (GalN)-sensitized mice. The aim of this study was to identify the role of Ron in the regulation of hepatic gene expression.
METHODS:Microarray analyses were performed on liver RNA isolated sequentially from wild-type (WT) and TK-/-mice during the progression of ALF. Gene array data were validated using Western and immunohistochemistry analyses as well as withex vivoculture systems.
RESULTS:At baseline, 101 genes were differentially expressed between WT and TK-/- livers, which regulate processes involved in hypoxia, proliferation, apoptosis and metabolism. One hour after ALF induction, WT livers exhibited increased cytokine expression compared to TK-/- livers, and after 4 hours, an induction of suppressor of cytokine signaling (SOCS) genes as well as JAK-STAT pathway activation were prominent in TK-/-livers compared to controls.
CONCLUSION:Our studies suggest a novel hepato-protective mechanism in Ron TK-/- mice wherein increased and sustained SOCS production and JAK-STAT activation in the hepatocyte may inhibit the destructive proinflammatory milieu and promote survival factors which blunt hepatic death and the ensuing development of ALF.
(Hepatobiliary Pancreat Dis Int 2012;11:383-392)
Mst1R; suppressors of cytokine signaling; Met receptor; hepatocyte growth factor-like protein
Introduction
Acute liver failure (ALF) affects about 2500 people in the United States annually with a mortality rate as high as 80% without a successful liver transplant.[1]The most common causes of ALF are drug toxicity (e.g. acetaminophen and idiosyncratic reactions), indeterminate liver disease, and viral hepatitis (e.g. hepatitis A and B).[1]Liver failure is heavily dependent on cytokine signaling and inflammation.[2]The major cell type mediating the production and release of proand anti-inflammatory cytokines and chemokines is the resident liver macrophage (Kupffer cells).[3-6]Although ALF has been studied for many years, little is known about the regulation of genes involved in the initial steps of promoting liver dysfunction and hepatocyte death.
The Ron receptor tyrosine kinase (TK) is a singlespanning membrane-bound heterodimeric glycoprotein that is expressed in most epithelial cells and on tissueresident macrophages.[7,8]Ron is a member of the Met family and has been implicated in a variety of biological processes including cancer progression and inflammation.[7-14]The ligand for Ron is the hepatocyte growth factor-like (HGFL) protein, also known as macrophage-stimulating protein.[15-17]HGFL binding induces Ron activation and the stimulation of downstream secondary messengers, which culminate to regulate changes in gene transcription.[7,8]
Mice lacking the TK signaling domain of Ron, referred to as Ron TK-/- mice,[18]are viable and have been used to study Ron functionin vivo. In a model of ALF induced by lipopolysaccharide (LPS) andD-galactosamine (GalN), Ron TK-/- mice display a hepato-protective phenotype compared to controls.[10]In this model, the TK-/- mice have mild aminotransferase elevation, minimal hepatocyte apoptosis, and relatively normal liver histology in contrast to wild-type (WT) mice. A recent study has further documented important cell-type-specific roles for Ron in regulating both Kupffer cell-dependent cytokine production and in inhibiting hepatocyte survival after injury.[19]
The goal of the present study was to identify Rondependent genes involved in the progression of ALF. To accomplish this, RNA was sequentially isolated from the livers of WT and Ron TK-/- mice after LPS/GalN treatment. Microarray assays were then used to define the genes that were altered between the genotypes during the progression of ALF. Our studies showed that the expression of suppressors of cytokine signaling (SOCS) genes that regulate cytokine secretion, as well as the JAK-STAT signaling pathway were increased in the TK-/- livers compared to controls. In total, our analyses demonstrated a potentially important mechanism by which Ron signaling influences the production of and response to cytokines during the progression of ALF.
Methods
Mice and endotoxin-induced ALF
Mice containing a germline deletion of the Ron TK signaling domain (TK-/- mice) have been previously described and were backcrossed onto a C57BL6 genetic background.[18,19]Wild-type (WT) and TK-/- mice were generated from heterozygous TK+/- crosses and then intercrossed to produce the experimental (TK-/-) and control (WT) mice. Male mice from 8 to 11 weeks were used. ALF was induced by intraperitoneal injection of LPS (27 ng/mg body weight; Sigma Chemical Co., St. Louis, MO) and GalN (1 mg/g body weight; Sigma) as previously described, and animals were sacrificed by carbon dioxide asphyxiation at the time points indicated.[10]
RNA extraction and microarray analysis
Total RNA was isolated from liver tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) as previously described.[19]RNA samples from two livers per genotype per time point were submitted to the Cincinnati Children's Hospital Medical Center Affymetrix Core. RNA quality was assessed and quantified using an Agilent Bioanalyzer 2100 (Hewlett Packard, Palo Alto, CA). Total RNA, 400-500 ng per sample, was used in the TargetAmp 1-Round Aminoallyl-aRNA amplification kit (Epicentre Biotechnologies, Madison, WI) to generate cRNA following the manufacturer's instructions. Biotin-X-X-NHS (Epicentre Biotechnologies, Madison, WI) was used to label the aminoallyl-aRNA with biotin. The biotin-labeled cRNA target was then chemically fragmented and hybridized to the Affymetrix MOE-430 GeneChip array containing about 39 000 targets. The probe arrays were scanned using the Affymetrix GeneChip Scanner 3000 and GeneChip Operating Software 1v4 (Affymetrix, Santa Clara, CA).
Data analysis
Changes in gene expression levels from the microarray signals were determined using algorithms in the Microarray Analysis Suite (Affymetrix) and GeneSpring GX 7.3.1 software (Silicon Genetics, Redwood City, CA). Gene lists from the microarray data were obtained based on a 1.5-fold expression difference using the Welchttest and 2-tailed Student'sttest (with or without Benjamini and Hochberg false discovery rate with FDR (P≤0.01). Correlation of gene expression with numeric parameters was assessed using the Pearson's product-moment correlation coefficient test withPvalue. Lists were filtered based on fold change andPvalue. Statistical significance among groups was determined using an unpaired 2-tail Welchttest or the Mann-WhitneyUtest with Bonferroni's correction. Gene clustering analyses were performed using GeneSpring GX 7.3 and K-means algorithms. The gene lists generated from the Gene Spring analyses were uploaded onto the DAVID Bioinformatics Resources 2008 National Institute of Allergy and Infectious Diseases, NIH website (http://david.abcc.ncifcrf.gov/) and pathway and functional annotation analyses were performed.
Kupffer cell and hepatocyte isolation and culture
Primary Kupffer cells and hepatocytes were isolated as previously described.[19]Kupffer cells were treated with bacterial LPS (500 ng/mL) and hepatocytes were pretreated with 200 ng/mL actinomycin D (ActD) followed by TNF-α (5 ng/mL) (R&D Systems, Minneapolis) for the times indicated.[19]
Quantitative real-time PCR
RNA isolation and cDNA preparation were performed as described previously.[19]Quantitative real-time PCR was performed using FastStart SYBR Green with an ABI 7900HT machine (Applied Biosystems, Foster City, CA). Expression levels were normalized to β-glucuronidase (GusB) as internal control. The experiments wererepeated twice with Kupffer cells pooled from 2 mice per isolation. Relative gene expression results are reported. The primers used were: GusB (5'-TTG AGA ACT GGT ATA AGA CGC ATC AG-3'; 5'-TCT GGT ACT CCT CAC TGA ACA TGC-3'), and SOCS3 (5'-TGG GCA GTG GGA GTG GTT ATT TCT-3'; 5'-ACT TTC CCG CTG GAA CTT GTT TGC-3').
Western blotting and immunohistochemistry
Western blotting was performed on the liver tissues and isolated hepatocytes as described previously.[19]The Western membranes from three independent experiments were quantified using ImageQuant software (GE Healthcare, Piscataway, NJ). Immunohistochemical analyses were performed on liver tissue sections as described previously.[19]The primary antibodies were phospho(p)- and total JAK2 (Millipore, Billerica, MA), pand total STAT-3 (Cell Signaling Technology, Danvers MA) and actin.
Results
We have previously shown that the absence of the Ron receptor TK diminishes the progression of ALF in mice; this finding was associated with dysregulation of cytokine production and decreased hepatocyte death through Ron regulation in both Kupffer cells and hepatocytes respectively.[10,19]Therefore, we hypothesized that Ron signaling would have a significant impact on the expression pattern of inflammatory and survival genes in the livers of injured mice. In this study, we proceeded to define the dysregulated gene expression profiles that potentially lead to the alteration of liver pathobiology between Ron-deficient (TK-/-) and WT animals.
Six Ron TK-/- and six WT control mice were injected with LPS and GalN, a proven liver-toxic regimen that initiates ALF in rodents.[3,10,20,21]Two animals per genotype were sacrificed at 0, 1 and 4 hours post-injection. Liver RNA was isolated and subjected to microarray analyses to identify differentially-expressed genes between WT and TK-/- livers basally and during the progression of ALF. We identified 101 genes that were differentially expressed between genotypes basally (Fig. 1A). The web-based DAVID gene function organizing tool[22]allowed grouping of most of these 101 genes into 45 overlapping biological process categories. Five categories from this list include response to hypoxia, cell proliferation, transcription, apoptosis and metabolism that account for two-thirds of the identified genes (Fig. 1B). Representative genes within these categories and their relative fold changes are indicated in Fig. 1C.

Fig. 1. 101 genes that were 1.5-fold changed in TK-/- livers compared to WT livers basally. A: Gene tree of all 101 dysregulated genes. B: Functional classification of a major subset of the genes based on biological process. Genes differentially expressed between untreated Ron TK-/- and WT mice were categorized using the DAVID GO term functional annotation tool. The number in parenthesis represents the total number of genes in those categories. C: Representative genes and their fold changes.
Two important components of the EGF signaling pathway were altered basally in the TK-/- livers compared to WT livers. The epidermal growth factor receptor (EGFR) and the janus kinase 1 (JAK1) genes were upregulated 2.5-fold and 1.8-fold respectively in TK-/-livers. These pathways have been shown to influence cytokine and chemokine levels, thereby promoting as well as diminishing inflammatory responses.[23,24]One of the most highly dysregulated genes basally was B-cell leukemia/lymphoma 6 (Bcl-6), which was expressed about 5-fold more in Ron TK-/- livers than in WT livers (Fig. 1C). The Bcl-6 gene encodes for a POZ/zinc finger transcriptional repressor that is ubiquitously expressed in adult mouse tissue and negatively regulates the expression of select cytokines.[25]A recent study has shown that Bcl-6 expression in pancreatic B-cells reduces cytokine induced-inflammation, Fas and iNOS expression as well as NO production, which protect against cytokineinduced apoptosis.[26]Similarly, higher Bcl-6 levels in the TK-/- liver may protect hepatocytes from cytokineinduced apoptosis in this LPS/GalN-induced ALF model.
Phosphatidylinositol 3-kinase (PI3K) is a key downstream mediator of many receptor TK signaling pathways. The alpha subunit of hypoxia inducible factor-1 (HIF-1α), one of the downstream targets of the PI3K pathway,[27]was basally high in Ron TK-/- deficient livers (Fig. 1C). HIF-1 transcription factor consists of α and β subunits but only the α subunit's expression is regulated whereas the β subunit is constitutively expressed.[27,28]HIF-1 encodes genes that respond to hypoxia. Three important functional categories of HIF-1α targets include cell proliferation, cell survival, and apoptosis.[27,28]It is possible that the high basal HIF-1α in TK-/- livers offers protection from ALF.
Upon LPS stimulation, Kupffer cells release a variety of inflammatory mediators including IL-6, IL-10, IFN-γ and TNF-α.[5,6,29]Correspondingly, the transcriptional levels of various cytokine and chemokine genes changed in the WT and TK-/- livers 1 hour after LPS/GalN injection (Table). The most highly upregulated cytokine gene at 1 hour was IL-6, showing a nearly 40-fold increase in mRNA levels in WT and TK-/- livers. IL-6 is one of the most prevalent inflammatory cytokines and mediates a multitude of effects in the cell including proliferation, survival and apoptosis.[30,31]However, the upregulation of IL-6 was very short-lived in WT and TK-/- livers. From 1 to 4 hours, the IL-6 expression returned to near baseline levels (Table). Another cytokine gene that was upregulated 1 hour after LPS/GalN treatment was TNF-α. TNF-α is required for the proper function of T, B and NK cells as well as macrophages including Kupffer cells.[32]TNF-α was upregulated 2.5-fold in WT livers comparedto 1.9-fold in TK-/- livers (Table), but returned to baseline levels after 4 hours in both genotypes. CXCL-2 (MIP-2) and CCL-3 (MIP-1α), chemokines involved in immune cell trafficking and function, were strongly expressed at 1 hour after ALF induction and the levels of each were approximately 2-fold higher in WT livers than in TK-/-livers (Table). From 1 to 4 hours after ALF induction, the CCL-3 level was not changed in WT livers, but was increased 1.8-fold in TK-/- livers.
In addition to cytokines, dramatic alterations in gene expression profiles of transcriptional regulators were observed between WT and TK-/- livers (Table). The expression of Jun oncogene was upregulated 2.8-fold inthe TK-/- livers compared to a 2.8-fold downregulation in WT livers 1 hour after ALF induction. The expression of Jun oncogene is controlled by cytokines and it also acts as a downstream mediator of cytokine signaling and controls the induction of SOCS genes.[33,34]Overall, 312 genes were differentially expressed (Fig. 2A, B) while 83 were altered to the same extent (Fig. 2C, D) in WT and TK-/- livers. After the use of K-means algorithms, the 312 differentially-expressed genes were clustered into 6 groups (Fig. 2B, D). Similar clustering was performed on the 83 genes regulated similarly in both genotypes. Amongst the differentially-regulated gene clusters, some exhibited increased expression in the WT livers, but decreased expression in the TK-/- livers, some showed decreased expression in the WT livers, but increased expression in the TK-/- livers, while others either showed increased or decreased expression in the genotypes, albeit to different extents, 1 hour after ALF induction (Fig. 2B, D).
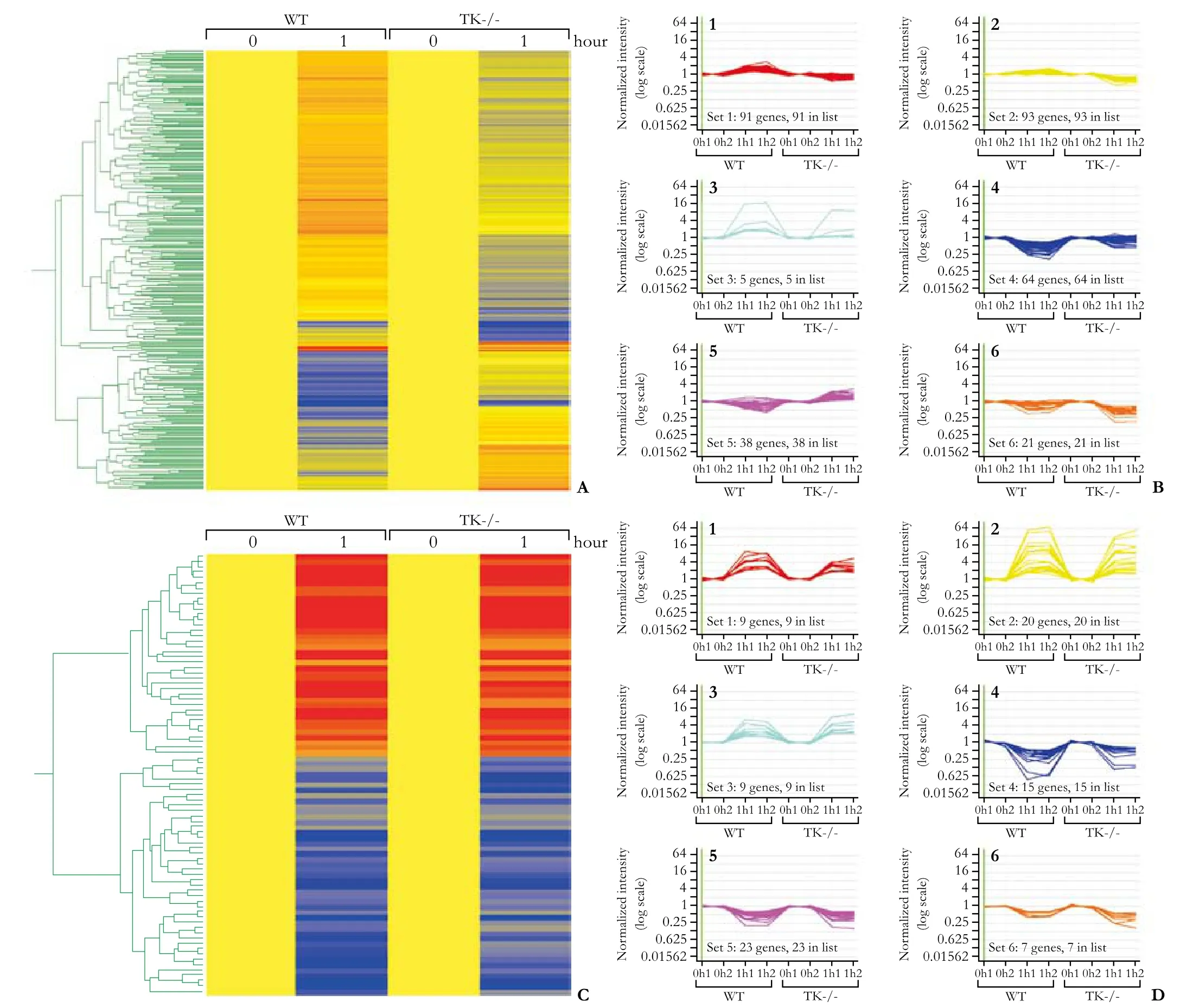
Fig. 2. Gene expression changes in WT and TK-/- livers after 1 hour of endotoxin and GalN exposure. A: 312 genes whose expression was altered differently at 1 hour after endotoxin exposure between WT and TK-/- livers. B: Cluster analysis of those 312 genes based on similarity of expression pattern. Using a K-means algorithm from GeneSpring, six clustering groups were established to identify patterns of gene expression. The X-axis shows the genotypes and treatment times while the Y-axis is the normalized intensity. C: 83 genes whose expression was altered similarly in both WT and TK-/- livers at 1 hour. D: Cluster analysis of 83 similarly regulated genes.
In our model, many cytokines, including interleukins such as IL-6, were highly upregulated within the first 60 minutes. These cytokines signal through the common janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, which can benegatively regulated by SOCS proteins.[35]SOCS gene expression is induced by various cytokines in a classical negative feedback loop. This inhibition occurs at the base of the signaling receptor by direct binding to proteins such as JAK2.[36]Further, the SOCS gene family members also inhibit the production of LPS-induced chemokines and cytokines.[37]Upregulation of SOCS genes was observed in the livers of WT and TK-/- mice within 1 hour after LPS/GalN exposure (Fig. 3). In this time-frame, SOCS1 was unchanged in WT animals, whereas SOCS2 expression was upregulated 2.7-fold and SOCS3 2.2-fold (Fig. 3A). In the livers of TK-/- mice, both SOCS1 and SOCS2 were unchanged while SOCS3 expression was upregulated 2.9-fold. However, from 1 to 4 hours of ALF induction, there was a significant difference in the expression levels of the SOCS 1, 2 and 3 genes between different genotypes (Fig. 3B). All three SOCS genes were upregulated in TK-/- livers, while only SOCS1 was upregulated in WT livers. In TK-/- livers, SOCS1 expression was increased by 3.5-fold, SOCS2 by 2.7-fold and SOCS3 by 1.9-fold. The study of SOCS genes may provide a mechanistic rationale for the protected phenotype observed in the TK-/- livers. Increased levels of all 3 SOCS genes may downregulate cytokine signaling pathways in TK-/- hepatocytes leading to a hepato-protective phenotype in Ron TK-/- mice.

Fig. 3. Suppressors of cytokine signaling were upregulated between 1 and 4 hours in the TK-/- animals whereas in the WT animals their expression levels returned to normal. The fold change of SOCS1, 2 and 3 expression was calculated at 0 to 1 hour (A) and at 1 to 4 hours (B) following LPS/GalN treatment. SOCS expression was maintained at high levels in the TK-/- livers compared to the livers from WT mice at the later time points, suggesting an enhanced downregulation of cytokine signaling in the TK-/-animals compared to controls.
To validate the microarray findings, Western analyses were performed on whole livers from WT and TK-/- mice 4 hours after LPS/GalN treatment. There was a significant increase of JAK2 level in the livers of TK-/- mice compared to WT mice (Fig. 4A, B). Immunohistochemical staining was performed on liver sections from LPS/GalN-treated mice 4 hours after ALF induction. The TK-/- livers showed stronger hepatocyte JAK2 staining than WT livers (Fig. 4C). Immunohistochemical analyses also demonstrated increased nuclear STAT3 staining in TK-/- hepatocytes compared to controls (Fig. 4C). To examine the impact of selective Ron loss in hepatocytes, primary hepatocytes were isolated from WT and TK-/- livers. Primary hepatocytes were subsequently treatedex vivowith actinomycin D (ActD) and TNF-α to induce an ALFresponse,[19]and cell lysates were isolated at the indicated time points (Fig. 4D). The increasing phosphorylation and total expression levels of JAK2 were analyzed by Western blotting in the TK-/- hepatocytes compared to controls (Fig. 4D).
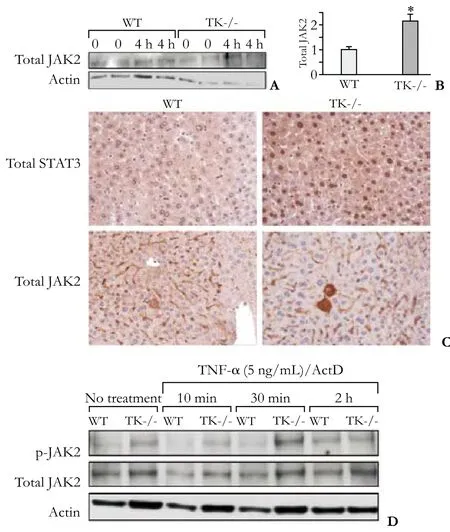
Fig. 4. Increased expression and activation of JAK2 and STAT3 in TK-/- liver and hepatocytes. A: Western analysis depicting increased JAK2 protein levels in TK-/- livers 4 hours after LPS/ GalN treatment. B: Densitometric quantification of Western blotting depicting increased JAK2 protein levels in TK-/- livers 4 hours after LPS/GalN treatment. *: P<0.05, compared to WT. C: Immunohistochemistry for STAT3 and JAK2 expression in liver sections from WT and TK-/- mice 4 hours after LPS/GalN treatment. Note the nuclear STAT3 localization in the TK-/-hepatocytes compared to controls (original magnification ×200). D: Ex vivo treatment of WT and TK-/- hepatocytes with TNF-α and ActD demonstrated increased p-JAK2 and total JAK2 levels in the TK-/- hepatocytes compared to controls.
To directly test if Ron activation regulates SOCS expression, Kupffer cells from WT and TK-/- mice were isolated and stimulated with LPSex vivo. TK-/-Kupffer cells exhibited exaggerated SOCS3 expression. No differences were observed between genotypes in the absence of LPS (Fig. 5). In total, our studies suggest a novel hepato-protective mechanism in TK-/- mice after LPS/GalN-induced liver failure. We propose that increased and/or temporally altered activation of the JAK-STAT-SOCS pathway in TK-/- hepatocytes promotes their survival and increased SOCS in TK-/-Kupffer cells blunts the production of an inflammatory cytokine milieu.
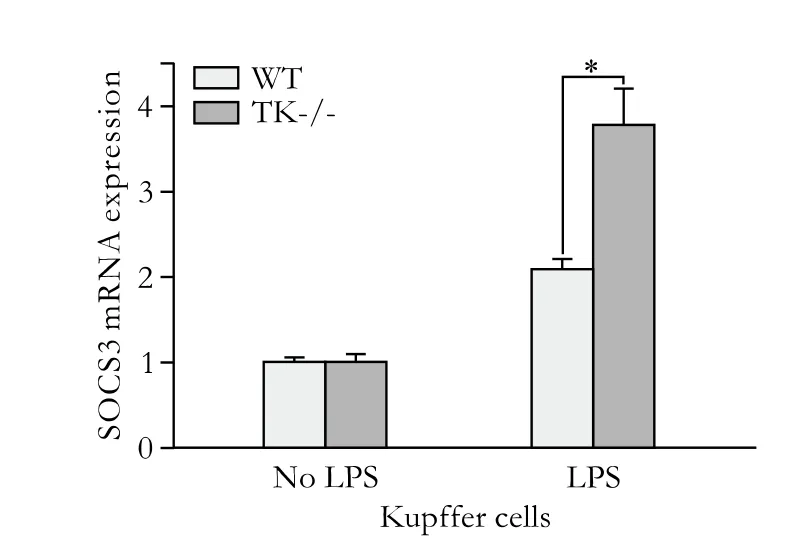
Fig. 5. SOCS3 was upregulated in TK-/- Kupffer cells more than WT cells after LPS stimulation. Quantitative real-time PCR analysis of LPS-stimulated Kupffer cells for the induction of SOCS3. TK-/-Kupffer cells displayed 1.8-fold more SOCS3 expression after LPS stimulation for 30 minutes than WT Kupffer cells. *: P<0.05.
Discussion
In the present study, we investigated the transcriptional regulation of genes induced in the liver of WT and Ron TK-/- mice during the progression of ALF modeled by endotoxin exposure. Microarray analyses were performed to define expression changes in any of the genes in the mouse genome. Our studies identified genes that were differentially expressed basally between WT and TK-/- livers as well as genes which were differentially regulated by Ron receptor signaling during ALF. Under basal conditions, 101 genes were different in Ron-replete and Ron-deficient livers. Genes that are basally regulated by Ron in the liver fall into 5 biological processes relating to the response to hypoxia, secretion, cell proliferation, cell death, and metabolism. The differential gene expression in these categories may be important for determining the response of the liver during injury.
Within 1 hour of exposure to LPS/GalN treatment, significant differential expression of 312 genes was noted in the livers of WT and TK-/- mice, while a number of other genes were also identified as being modulated during ALF but did not differ between genotypes. Of the genes induced during early injury, numerous cytokine and chemokine gene expressions were dysregulated to varying degrees. Dramatic upregulation of IL-6 was observed in both genotypes while the levels of CXCL2 (MIP-2) and CCL-3 (MIP-1α) were more highly (2-fold) expressed in WT livers versus TK-/- livers. Increasing levels of CXCL-2 and CCL-3 may help attract leukocytes contributing to the hepatic inflammatory environment.[38,39]These data are consistent with our previous observation showing that WT livers display increased neutrophil recruitment compared toTK-/- livers.[10]Although recruitment of leukocytes is a key component of the immune response and hostdefense mechanism, in various disease conditions, including endotoxemia, the activation and infiltration of leukocytes may also be a factor leading to increased tissue damage in WT livers.[40]

Fig. 6. Working model. Based on our data, a working model for the hepato-protective phenotype observed in Ron TK-/- livers of mice exposed to LPS/GalN was generated. We propose that high levels of SOCS genes during ALF seen in TK-/- liver antagonize the LPS-induced expression and function of inflammatory cytokines/chemokines in Kupffer cells as well as antagonize the deleterious effect of cytokine signaling in the TK-/- hepatocytes. High levels of SOCS and Bcl-6 as well as higher NF-κB activity in TK-/- livers may also contribute to hepato-protection.
Three members of the SOCS gene family (SOCS1, SOCS2 and SOCS3) were differentially regulated in the WT and TK-/- livers. SOCS proteins are induced by cytokines and act in a negative feedback loop to inhibit cytokine gene expression. SOCS1, 2 and 3 exhibited increased and prolonged expression in the TK-/- livers compared to controls. Given the high expression of IL-6R in hepatocytes,[41]it is interesting to speculate that IL-6 signaling varies between the WT and Ron TK-/-livers. Elevated IL-6 during ALF in Ron TK-/- livers leads to increases in SOCS expression, blocking the deleterious effects of cytokine signaling in the TK-/-hepatocyte.
Analyses of liver tissues showed that the JAK2-STAT3 pathway was hyperactivated in the TK-/- livers compared to control livers. Further, recent studies in mice have shown that the JAK2/STAT3 pathway initiates survival signals and that the activation of this pathway in hepatocytes is essential to protect these cells from LPS/GalN-induced apoptosis.[42]SOCS proteins, by themselves, also activate survival/anti-apoptotic pathways in various cell types.[35]
In the liver, Kupffer cells are the major cell type responding to LPS, resulting in a robust increase in cytokine chemokine secretion.[5,6,29]LPS binds to and activates toll-like receptor 4 (TLR-4), initiating a signaling cascade that results in NF-κB phosphorylation and activation of cytokine chemokine secretion.[5,6,29]SOCS3 has been shown to inhibit LPS-induced cytokine chemokine secretion by interacting with the TLR-4 pathway.[43,44]SOCS3 is upregulated by LPS in macrophages and acts in a negative feedback loop to attenuate inflammatory responses. Prior studies have shown that intracellular delivery of a recombinant cell-penetrating form of SOCS3 protein in different mouse models of ALF, including the LPS/GalN model, significantly increase survival and decrease inflammatory responses with reduction in hepatic hemorrhage and necrosis.[45]Similarly, we speculate that overexpressing SOCS3 specifically in the WT hepatocytes may make them resistant to LPS/GalN-induced death, similar to the TK-/- hepatocytes. Increasing SOCS3 in the WT Kupffer cells may similarly decrease the high levels of cytokines found after LPS/ GalN treatment. Increased levels of SOCS3 seen in TK-/- Kupffer cells over time may alter the nature of the inflammatory cytokine/chemokine milieu, making it less toxic.
Based on the combined data, Fig. 6 outlines a proposed model for the role of Ron signaling in regulating ALF. In this model, Kupffer cell activation (through LPS stimulation) leads to the production of cytokines and chemokines. In Kupffer cells, loss of Ron signaling may lead to increasing SOCS expression, which may alter cytokine gene expression. Upregulation of the JAK2/STAT3 signaling pathway in TK-/- hepatocytes alters the response of these cells to the cytokine and chemokine milieu. Hyperactivation of the JAK2/STAT3 pathway, along with increases in pro-survival genes such as Bcl-6 in TK-/- livers may confer hepato-protective effects after LPS/GalN-induced ALF. This implies that Ron receptor signaling in Kupffer cells as well as hepatocytes regulates the expression of SOCS genes. Further, SOCS induction in Kupffer cells and hepatocytes in TK-/- livers plays a role in resistance to ALF.
In summary, this report has identified a novel signaling mechanism by which the Ron receptor regulates ALF progression that may be used to design targeted therapeutic interventions for decreasing the morbidity and mortality associated with this disease.
Contributors:KRM, KLW, CDJ and LMA were involved in the design, acquisition and analysis of the data as well as in drafting the manuscript. SWD was involved in the acquisition of data and editing the manuscript. WSE was involved in the conception and design of the study as well as in drafting and revising the manuscript. WSE is the guarantor.
Funding:This work was supported by grants from the Public Health Services DK-73552 (WSE), and the Digestive Diseases Research Development Center DK-064403 (WSE and LMA) from the National Institutes of Health, as well as by grant project #8950 (WSE) from Shriner's Hospital for Children.
Ethical approval:All procedures were approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Competing interest:The authors do not choose to declare any conflict of interest related directly or indirectly to the subject of this article.
1 Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol 2009;6:542-553.
2 Igonin AA, Armstrong VW, Shipkova M, Lazareva NB, Kukes VG, Oellerich M. Circulating cytokines as markers of systemic inflammatory response in severe communityacquired pneumonia. Clin Biochem 2004;37:204-209.
3 Galanos C, Freudenberg MA, Reutter W. Galactosamineinduced sensitization to the lethal effects of endotoxin. Proc Natl Acad Sci U S A 1979;76:5939-5943.
4 Hishinuma I, Nagakawa J, Hirota K, Miyamoto K, Tsukidate K, Yamanaka T, et al. Involvement of tumor necrosis factor-alpha in development of hepatic injury in galactosaminesensitized mice. Hepatology 1990;12:1187-1191.
5 Jirillo E, Caccavo D, Magrone T, Piccigallo E, Amati L, Lembo A, et al. The role of the liver in the response to LPS: experimental and clinical findings. J Endotoxin Res 2002;8: 319-327.
6 Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48:322-335.
7 Leonis MA, Thobe MN, Waltz SE. Ron-receptor tyrosine kinase in tumorigenesis and metastasis. Future Oncol 2007;3: 441-448.
8 Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res 2008;100:1-33.
9 Caldwell CC, Martignoni A, Leonis MA, Ondiveeran HK, Fox-Robichaud AE, Waltz SE. Ron receptor tyrosine kinasedependent hepatic neutrophil recruitment and survival benefit in a murine model of bacterial peritonitis. Crit Care Med 2008;36:1585-1593.
10 Leonis MA, Toney-Earley K, Degen SJ, Waltz SE. Deletion of the Ron receptor tyrosine kinase domain in mice provides protection from endotoxin-induced acute liver failure. Hepatology 2002;36:1053-1060.
11 Mallakin A, Kutcher LW, McDowell SA, Kong S, Schuster R, Lentsch AB, et al. Gene expression profiles of Mst1r-deficient mice during nickel-induced acute lung injury. Am J Respir Cell Mol Biol 2006;34:15-27.
12 McDowell SA, Mallakin A, Bachurski CJ, Toney-Earley K, Prows DR, Bruno T, et al. The role of the receptor tyrosine kinase Ron in nickel-induced acute lung injury. Am J Respir Cell Mol Biol 2002;26:99-104.
13 Tsutsui S, Noorbakhsh F, Sullivan A, Henderson AJ, Warren K, Toney-Earley K, et al. RON-regulated innate immunity is protective in an animal model of multiple sclerosis. Ann Neurol 2005;57:883-895.
14 Wetzel CC, Leonis MA, Dent A, Olson MA, Longmeier AM, Ney PA, et al. Short-form Ron receptor is required for normal IFN-gamma production in concanavalin A-induced acute liver injury. Am J Physiol Gastrointest Liver Physiol 2007;292: G253-261.
15 Bezerra JA, Witte DP, Aronow BJ, Degen SJ. Hepatocytespecific expression of the mouse hepatocyte growth factorlike protein. Hepatology 1993;18:394-399.
16 Gaudino G, Follenzi A, Naldini L, Collesi C, Santoro M, Gallo KA, et al. RON is a heterodimeric tyrosine kinase receptor activated by the HGF homologue MSP. EMBO J 1994;13:3524-3532.
17 Wang MH, Ronsin C, Gesnel MC, Coupey L, Skeel A, Leonard EJ, et al. Identification of the ron gene product as the receptor for the human macrophage stimulating protein. Science 1994;266:117-119.
18 Waltz SE, Eaton L, Toney-Earley K, Hess KA, Peace BE, Ihlendorf JR, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J Clin Invest 2001;108:567-576.
19 Stuart WD, Kulkarni RM, Gray JK, Vasiliauskas J, Leonis MA, Waltz SE. Ron receptor regulates Kupffer cell-dependent cytokine production and hepatocyte survival following endotoxin exposure in mice. Hepatology 2011;53:1618-1628.
20 Decker K, Keppler D. Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev Physiol Biochem Pharmacol 1974;77-106.
21 Konishi Y, Shinozuka H, Farber JL. The inhibition of rat liver nuclear ribonucleic acid synthesis by galactosamine and its reversal by uridine. Lab Invest 1974;30:751-756.
22 Delker DA, Geter DR, Roop BC, Ward WO, Ahlborn GJ, Allen JW, et al. Oncogene expression profiles in K6/ODC mouse skin and papillomas following a chronic exposure to monomethylarsonous acid. J Biochem Mol Toxicol 2009;23: 406-418.
23 Hamilton LM, Torres-Lozano C, Puddicombe SM, Richter A, Kimber I, Dearman RJ, et al. The role of the epidermal growth factor receptor in sustaining neutrophil inflammation in severe asthma. Clin Exp Allergy 2003;33:233-240.
24 Rocourt DV, Mehta VB, Besner GE. Heparin-binding EGF-like growth factor decreases inflammatory cytokine expression after intestinal ischemia/reperfusion injury. J Surg Res 2007; 139:269-273.
25 Jardin F, Ruminy P, Bastard C, Tilly H. The BCL6 protooncogene: a leading role during germinal center development and lymphomagenesis. Pathol Biol (Paris) 2007;55:73-83.
26 Igoillo-Esteve M, Gurzov EN, Eizirik DL, Cnop M. The transcription factor B-cell lymphoma (BCL)-6 modulates pancreatic {beta}-cell inflammatory responses. Endocrinology 2011;152:447-456.
27 Hellwig-Bürgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res 2005;25:297-310.
28 Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3:721-732.
29 Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol 2002;283:G256-265.
30 Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 2005;41:2502-2512.
31 Hodge DR, Peng B, Cherry JC, Hurt EM, Fox SD, Kelley JA, et al. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res 2005;65: 4673-4682.
32 Aggarwal BB, Shishodia S, Takada Y, Jackson-Bernitsas D, Ahn KS, Sethi G, et al. TNF blockade: an inflammatory issue. Ernst Schering Res Found Workshop 2006:161-186.
33 Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem 1996;271:15703-15707.
34 Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharideinduced SOCS-3 gene expression in macrophages and microglia. J Immunol 2007;179:5966-5976.
35 Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007;7: 454-465.
36 Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci 2004;117:1281-1283.
37 Strengell M, Lehtonen A, Matikainen S, Julkunen I. IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J Leukoc Biol 2006;79:1279-1285.
38 Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA. Role for macrophage inflammatory protein-2 in lipopolysaccharideinduced lung injury in rats. J Immunol 1996;156:1963-1972.
39 Shanley TP, Schmal H, Friedl HP, Jones ML, Ward PA. Role of macrophage inflammatory protein-1 alpha (MIP-1 alpha) in acute lung injury in rats. J Immunol 1995;154:4793-4802.
40 Jaeschke H. Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemiareperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol 2006;290:G1083-1088.
41 Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997;276:60-66.
42 Wang K, Damjanov I, Wan YJ. The protective role of pregnane X receptor in lipopolysaccharide/D-galactosamineinduced acute liver injury. Lab Invest 2010;90:257-265.
43 Kinjyo I, Hanada T, Inagaki-Ohara K, Mori H, Aki D, Ohishi M, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity 2002;17:583-591.
44 Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity 2002;17:677-687.
45 Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med 2005;11:892-898.
Accepted after revision March 14, 2012
The purpose of knowledge is to liberate the person from restrictive limits while simultaneously maintain contact with the perceptible and ponderable universe.
—Wade Nobles
December 29, 2011
Author Affiliations: Departments of Cancer and Cell Biology (Kulkarni RM, Stuart WD and Waltz SE) and Biology (Kutcher LW and Carson DJ), University of Cincinnati, Cincinnati, OH 45267-0521, USA; Division of Gastroenterology, Hepatology and Nutrition, Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229, USA (Leonis MA); Department of Research, Shriner's Hospital for Children and Cincinnati Veterans Affairs Medical Center, Cincinnati, OH 45267-0521, USA (Waltz SE)
Susan E Waltz, PhD, Department of Cancer and Cell Biology, 3125 Eden Ave., University of Cincinnati College of Medicine, Cincinnati, OH 45267-0521, USA (Tel: 513-558-8675; Email: susan.waltz@ uc.edu)
© 2012, Hepatobiliary Pancreat Dis Int. All rights reserved.
10.1016/S1499-3872(12)60196-9
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Gastric- and intestinal-type marker expression in invasive ductal adenocarcinoma of the pancreas
- Early changes of hepatic hemodynamics measured by functional CT perfusion in a rabbit model of liver tumor
- A common variant in the precursor miR-146a sequence does not predispose to cholangiocarcinoma in a large European cohort
- Muscarinic acetylcholine receptor M3 in proliferation and perineural invasion of cholangiocarcinoma cells
- Hepatocyte differentiation of mesenchymal stem cells
- Early control of short hepatic portal veins in isolated or combined hepatic caudate lobectomy