鸡毒支原体磷酸酶PrpC活性检测
2012-06-29胡美容陈鸿军于圣青仇旭升
胡美容,陈鸿军,于圣青,仇旭升,谭 磊,高 崧,丁 铲*
(1.中国农业科学院上海兽医研究所,上海 200241;2.扬州大学兽医学院,江苏 扬州 225009)
鸡毒支原体(Mycoplasma gallisepticum,MG)能引发鸡慢性呼吸道疾病,主要表现为呼吸啰音,咳嗽,流鼻涕,严重时可见呼吸困难[1]。为适应营寄生生活,MG通过调节黏附素的生物学活性来适应外界环境的变化,以控制支原体入侵细胞、从细胞膜表面脱离或从细胞内出芽释放等过程[2]。
由于支原体基因组结构极为简单,其在转录水平上的基因调控子十分有限,且普遍缺乏二元信号调节系统[3]。在肺炎支原体(M.pneumoniae)中,转录调节因子仅占0.5%,仅存在单一的Sigma因子和3种转录阻遏蛋白[4]。近年来的研究发现,支原体具备蛋白翻译后修饰功能,以蛋白磷酸化占主导[5]。其中,研究最多的是磷酸糖转移酶系统(PTS)。该系统包括酶I、HPr和葡萄糖渗透酶[6]。酶I对HPr的H15位点进行磷酸化修饰,支原体可借助该系统将磷酸烯醇式丙酮酸的磷酸基团转移至底物[7]。
研究发现,肺炎支原体HPr S46位点亦可被磷酸化修饰,而催化酶为HPr激酶(PrkC),该过程需要ATP参与[8]。这是抑制碳降解途径关键性修饰过程,其逆过程由磷酸酶 PrpC(mpn247)催化[9]。PrpC是一种PPM磷酸酶(即对Mn2+或Mg2+依赖的蛋白磷酸酶),可对PrkC的自身磷酸化修饰位点进行去磷酸化[10]。prpC与prkC同在一个基因簇上,部分基因序列重叠,含有相同的启动元件,表达时相一致[11]。PrkC/PrpC系统在细菌和支原体中均高度保守,不仅参与糖酵解酶类的磷酸化/去磷酸化,而且严重影响着生物被膜和芽胞形成、毒力和分裂生殖等重要的生命活动[12]。本文克隆并突变获得编码鸡毒支原体PrpC磷酸酶的ptc1基因,对原核表达产物的酶活性进行细致分析,旨在建立PrpC功能检测体系,为进一步深入研究PrkC/PrpC调节系统奠定基础。
1 材料与方法
1.1 材料
鸡毒支原体标准强毒株Rlow、大肠埃希菌DH5α和BL21(DE3)工程菌均为本实验室保存。
pGEM-T Easy载体,T4DNA连接酶均购自美国Promega公司;pET-28a载体,购自 Merck公司;Platinum®Taq DNA Polymerase High Fidelity,购自Invitrogen公司;质粒DNA提取试剂盒,凝胶回收DNA试剂盒,购自Qiagen公司;碱性磷酸酶(Alkaline Phosphatase,CIAP)、BamHⅠ、EcoRⅠ、XhoⅠ等,购自宝生物工程(大连)有限公司;1kb DNA Marker和小分子质量蛋白预染Marker,购自MBI公司;支原体培养用Bacto heart infusion broth、Agar noble,购自BD公司;灭活马血清和猪血清,购自Gibco公司;细菌培养用Tryptone、Yeast Extract,购自OXOID公司;His Band Resin蛋白纯化试剂盒,购自Novagen公司;HRP标记羊抗鼠IgG 和对硝基苯 (p-NitropHenyl Phosphate Liquid Substrate System,PNPP)脂磷酸盐溶液,购自Sigma公司。
1.2 方法
1.2.1 鸡毒支原体DNA提取及PCR扩增 鸡毒支原体Rlow株生长于ATCC完全培养基中(100 mL/L灭活马血清+100mL/L的灭活猪血清+100 mL/L新鲜酵母提取物),置37℃、体积分数为5%的CO2培养箱静置培养3d或摇床中培养16h至对数生长期。取2mL于EP管中,12000r/min离心10min,沉淀用无菌PBS洗涤3次,最后用50μL无菌PBS悬浮,煮沸10min后,冰浴5min,12000 r/min离心10min,取上清,测定DNA含量。
由于ptc1基因中含有1个编码色氨酸的TGA三联子,该三联子在大肠埃希菌中为终止子,为了在大肠埃希菌中有效表达PrpC蛋白全长序列,设计一对中间突变引物ptc1R(AGTCTCCCACCCAGA ACGTGT)和 ptc2F (ACACGTTCTGGGTGGGAGACT)突变该位点为TGG。取100pg Rlow基因组 DNA 为模板,用PlatinumⓇTaq DNA Polymerase High Fidelity分两段扩增,ptc1基因前段(ptc1N)用引物ptc1F (CAAGGATCCATGAAAAAGATATATGCAAGTTCGAC)和ptc1R;用ptc2F和ptc2R(CAAGAATTCTTAGCCATTAATAACCTCAGTTAG)扩增ptc1基因后半段(ptc1C),反应条件均为:94℃预变性2min;94℃30s,55℃30s,68℃1min,共30个循环;68℃延伸10min。将ptc1N和ptc1CPCR产物分别回收,各取10ng作为模板,利用ptc1F和ptc2R引物扩增ptc1基因全长,反应条件为:94℃预变性2 min;94℃30s,55℃1min,68℃2min,共30个循环;68℃延伸10min。PCR产物经纯化后,与pGEM T-easy载体连接,转化E.coli DH5α,进行蓝白斑筛选,挑取克隆酶切鉴定,取阳性质粒送上海Invitrogen公司测序。
1.2.2 PrpC原核表达及纯化 将测序正确的阳性质粒经BamHⅠ、EcoRⅠ酶切后,与同样酶切的pET-28a(+)载体连接,转化DH5α,挑菌,提质粒酶切鉴定,将阳性质粒转BL21(DE3),用0.5mmol/L IPTG进行诱导表达,超声波裂解(400W,工作时间5s,间歇时间5s,99次),彻底破碎细菌,12000 r/min离心15min,收集上清和包涵体。按常规方法[5]制备蛋白样品,进行SDS-PAGE电泳鉴定,用His Band Resin试剂盒纯化表达产物,BCA试剂盒定量蛋白含量。
1.2.3 PrpC酶活性检测
1.2.3.1 比活性测定 在荧光酶标板中加入192 μL PNPP反应底物,调节 Mn2+浓度至2mmol/L,总体积调整至200μL,每孔加入不同浓度Ptc1表达产物(1.5、3、4.5、6、7.5、9μg),混匀,37℃放置5 min后,测定OD405nm吸光度值。
1.2.3.2 最佳 Mn2+浓度确定 在EP中加入192 μL PNPP反应底物,然后依次加入不同浓度的Mn2+(0、0.2、0.3、0.4、0.5、1、2、3、4、5mmol/L),总体积调整至200μL,加入6μg Ptc1表达产物,37℃作用5min后,在多功能酶标仪中测定OD405nm吸光度值。
1.2.3.3 最适pH 确定 用 HCl/NaOH 调节酶底物反应液(192μL PNPP,2mmol/L Mn2+/8μL)的pH,设定pH从1到12,再加入6μg Ptc1表达产物,37℃作用5min后,在多功能酶标仪中测定OD 405nm吸光度值。
1.2.3.4 最适温度确定 在不同EP管中加入200 μL酶底物反应液(Mn2+终浓度为2mmol/L),置于不同温度下预冷或预热(0、25、37、45、50、55、60℃)5 min,于各管中加入等量6μg Ptc1表达产物,混匀后,置于不同温度下作用5min,分别测定其吸光度值。
1.2.3.5 不同金属离子对酶活性影响 在不同EP管中加入200μL PNPP 反应底物(2mmol/L Mn2+,pH8.5),然后依次加入相同浓度的 LiCl、NaCl、KCl、MgCl2、CaCl2、BaCl2、HgCl2、AlCl3、MnCl2溶液,混匀后,加入6μg Ptc1表达产物,37℃反应5min,分别测定其吸光度值。
2 结果
2.1 PrpC原核表达及纯化
通过Overlap PCR扩增获得含点突变的ptc1基因。将该基因亚克隆入pET-28a(+)载体中,阳性质粒命名为pET-ptc1。用IPTG诱导转化有pET-ptc1重组质粒的 BL21(DE3),经SDS-PAGE鉴定,该重组蛋白呈现可溶性表达,经 His Band Resin试剂盒纯化,获得2mg重组蛋白,溶于1mL Elution buffer中(图1)。
2.2 PrpC比活性测定
为了检测可溶性表达Ptc1蛋白在体外是否具有酶活性,本实验以CIAP为阳性对照,反应底物均为200μL PNPP溶液(含2μL终浓度为2mmol/L Mn2+),CIAP反应孔则加入等量蒸馏水。结果可见,纯化的Ptc1蛋白具有酶反应活性。在相同条件下,Ptc1蛋白的OD值高于CIAP(图2)。
2.3 最适Mn2+浓度及PrpC酶反应浓度的摸索
Mn2+在0~2mmol/L范围内,随着浓度的增加,PrpC酶反应活性逐渐增强;当 Mn2+≥2 mmol/L后,Ptc1蛋白的反应活性维持在平台期(图3A)。因此,本试验选2mmol/L Mn2+为PrpC最佳离子反应浓度。随后,在底物浓度不变的情况下,摸索PrpC的最适反应酶量,结果显示,在200μL PNPP及2mmol/L Mn2+的底物反应液中,Ptc1蛋白最适酶量为6μg(图3B)。
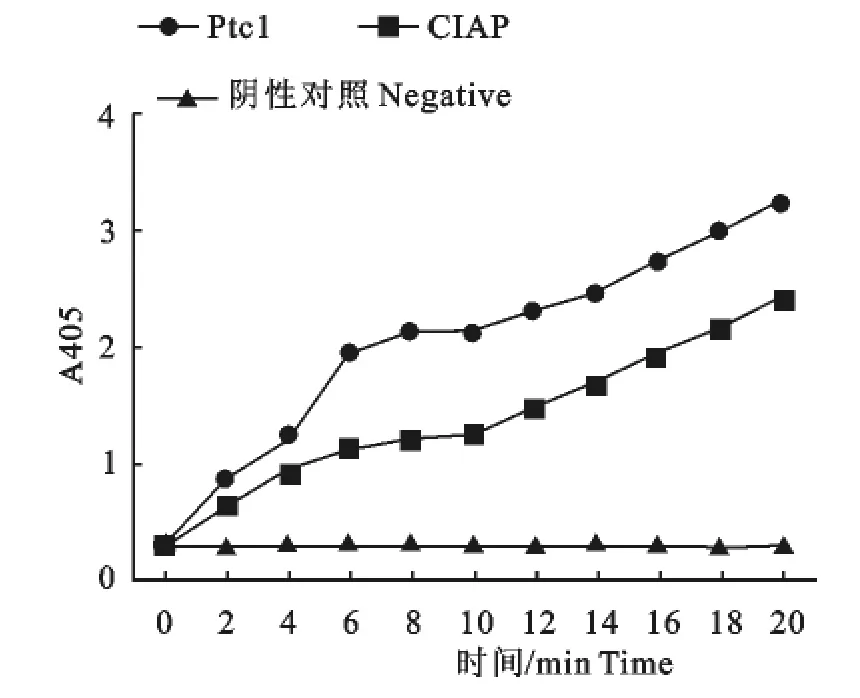
图2 Ptc1蛋白比活性测定Fig.2 Ptc1enzyme activity was determined

图3 最适Mn2+浓度及PrpC酶反应浓度Fig.3 The determination of optimum concentrations of Mn2+ and PrpC
2.4 最佳pH、最适温度的确定
在确定Mn2+反应浓度及PrpC酶反应量后,在反应体系中(200μL PNPP,2mmol/L Mn2+,6μg Ptc1蛋白)测定了PrpC的最适pH(图4A)和最适反应温度(图4B)。结果表明:PrpC的最适反应pH为8.5,该酶为碱性磷酸酶;最适反应温度为37℃。

图4 最佳反应温度和pH确定Fig.4 The determination of optimum reaction temperature and pH
2.5 不同金属离子对PrpC酶活性的影响
PrpC在无Mn2+存在下,即使加入其他金属离子,不具有酶活性。因此,在检测不同金属离子对PrpC的影响时,底物反应液中均加入2mmol/L Mn2+。结果显示,Ba2+对Ptc1蛋白酶活性的影响最小,其相对活性为78.2%,Li+、Na+、Ca2+、Mg2+次之,K+、Al3+对Ptc1蛋白酶活性有较大干扰,Hg2+能完全抑制PrpC(表1)。

表1 不同金属离子对PrpC酶活性影响Table1 Effects of metal irons on PrpC activity
3 讨论
在原核和真核生物中,蛋白可逆的磷酸化作用起着关键作用,如代谢途径调节,细胞分化以及信号转导[13,7,3]。在真核生物体内,丝氨酸/苏氨酸蛋白的磷酸酶类根据其结构、金属离子依赖性以及对抑制剂的敏感程度等不同可分为两大类,即PPP和PPM[14-15]。序列比较发现,这两大类磷酸酶有一个共同的催化中心,即PPP的第220位氨基酸和PPM 的第290位氨基酸[15,8]。PPM 类磷酸酶的特征是在其序列中,连续或间接地有11个保守基序,其中8个基序绝对保守。PPM类磷酸酶家族中人类的PP2C最具代表性。PP2C样酶依赖Mg2+或Mn2+来催化磷酸丝氨酸和磷酸苏氨酸残基的去磷酸化作用[9]。PrpC是PP2C蛋白磷酸酶家族中的一员。这类酶为Mn2+或Mg2+依赖性,其磷酸化底物非常广泛,包括人工合成的对硝基苯脂磷酸盐溶液(PNPP),蓝细菌的PⅡ蛋白[16],或者是枯草杆菌的抗σ因子和翻译延伸因子EF-G[13,17]。其中,PNPP底物在磷酸化状态时为无色,当其被去磷酸化后溶液则变为黄色,底物的颜色变化可通过多功能酶标仪在OD405nm处测定其吸光值。
本文选择鸡毒支原体PrpC(ptc1基因编码)进行克隆、表达和纯化,并对其酶反应特性进行分析。原核表达的PrpC蛋白在体外能成功地将人工合成的磷酸化底物PNPP去磷酸化,该酶为Mn2+依赖型,当 Mn2+终浓度≥2mmol/L时,PrpC的活性最强;该酶的最适反应pH为8.5,最适反应温度是37℃。1mmol/L 的 Li+、Na+、K+、Mg2+、Ca2+、Ba2+、Al3+、Hg2+等金属离子在2mmol/L Mn2+存在下,对PrpC均有一定的抑制作用,其中Hg2+能完全抑制其活性,在K+、Al3+离子存在时,PrpC的酶活性仅为20%作用,而在 Li+、Na+、Ca2+、Mg2+存在下,也只有约30%的活性,Ba2+对该酶的影响最小,在Ba2+存在下,该酶还能保留78.2%的活性。这些试验结果与Obuchowski M等观察到枯草杆菌中PrpC的蛋白特性结果相似[7]。
Sebastian R S等[3]绘制了肺炎支原体磷酸化蛋白表达谱。他们通过磷酸多肽富集结合质谱分析发现,肺炎支原体中存在63种磷酸化蛋白,占总蛋白的近10%。P1、P30、HMW1、HMW2、HMW3等黏附素、表面未鉴定蛋白MPN474及一些代谢酶类(如烯醇化酶)均被证实可以被磷酸化。同时,他们通过Tn4001转座子缺失筛选获得了PrkC缺失株,并通过黏附素磷酸化修饰的Pro-Q染色-质谱检测,发现了PrkC缺失株中,所有上述蛋白的磷酸化修饰丧失,与此同时,致病力亦丧失;PrkC可直接作用HMW3、P41、MPN474、MPN256、P1和HMW1等,磷酸化后修饰的HMW1和HMW3增加了与P1和表面MPN474的组装稳定性,PrkC缺失后,黏附蛋白解聚,不再定位于顶突中[18]。
在MG中,被鉴定的磷酸化修饰黏附相关蛋白极少。Demina I A等[2]通过磷酸多肽富集技术结合2-DIGE技术,对 MG S6株可溶性蛋白进行鉴定。结果发现,在可溶的466种胞浆蛋白中,有1.5%的功能蛋白可被磷酸化修饰,主要是分子伴侣和翻译调控蛋白,如 TufB、MalK/PotA、GroEL/Hsp60、DnaK、Efp等,仅有一种含量极高的血凝素/黏附素VlhA(LP64)被检测出含磷酸化位点。在这些功能蛋白的磷酸化修饰过程中,PrkC/PrpC系统扮演着怎样的角色?该系统是否参与调控鸡毒支原体细胞黏附活动关键蛋白的磷酸化修饰过程?这些均尚未证实。因此,对PrpC功能的研究对于鸡毒支原体致病性研究具有重要价值。而这需要基于下一步构建MG PrpC定点缺失株,对其生物学特性的变化来证实。
[1]Nunoya T,Kanai K,Yagihashi T,et al.Natural case of salpingitis apparently caused by Mycoplasma gallisepticumin chickens[J].Avian Pathol,1997,26:391-398.
[2]Demina I A,Serebryakova M V,Ladygina V G,et al.Proteome of the bacterium Mycoplasma gallisepticum [J].Biochemistry(Mosc),2009;74:165-174.
[3]Schmidl S R,Gronau K,Hames C,et al.The stability of cytadherence proteins in Mycoplasma pneumoniae requires activity of the protein kinase PrkC[J].Infect Immun,2010,78:184-192.
[4]Loens K,Beck T,Ursi D,et al.Development of real-time multiplex nucleic acid sequence-based amplification for detection of Mycoplasma pneumoniae,Chlamydophila pneumoniae,and Legionella spp.in respiratory specimens[J].J Clin Microbiol,2008,46:185-191.
[5]Seil R,Bossers T,Kuhn P,et al.Rev Chir Orthop Reparatrice Appar Mot[J].Pub Med,2005,91:56.
[6]Postma P W,Lengeler J W,Jacobson G R.Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria[J].Microbiol Rev,1993,57:543-594.
[7]Halbedel S,Busse J,Schmidl S R,et al.Regulatory protein phosphorylation in Mycoplasma pneumoniae.A PP2C-type phosphatase serves to dephosphorylate HPr(Ser-P)[J].J Biol Chem,2006,281:26253-26259.
[8]Stulke J,Arnaud M,Rapoport G,et al.PRD--aprotein domain involved in PTS-dependent induction and carbon catabolite repression of catabolic operons in bacteria[J].Mol Microbiol,1998,28:865-874.
[9]Darbon E,Servant P,Poncet S,et al.Antitermination by GlpP,catabolite repression via CcpA and inducer exclusion triggered by P-GlpK dephosphorylation control Bacillus subtilis glpFK expression[J].Mol Microbiol,2002,43:1039-1052.
[10]Obuchowski M,Madec E,Delattre D,et al.Characterization of PrpC fromBacillus subtilis,a member of the PPM phosphatase family[J].J Bacteriol,2000,182:5634-5638.
[11]Iwanicki A,Hinc K,Seror S,et al.Transcription in theprpC-yloQ region in Bacillus subtilis [J].Arch Microbiol,2005,183:421-430.
[12]Madec E,Laszkiewicz A,Iwanicki A,et al.Characterization of a membrane-linked Ser/Thr protein kinase in Bacillus subtilis,implicated in developmental processes[J].Mol Microbiol,2002,46:571-586.
[13]Adler E,Donella-Deana A,Arigoni F,et al.Structural relationship between a bacterial developmental protein and eukaryotic PP2Cprotein phosphatases[J].Mol Microbiol,1997,23:57-62.
[14]Deutscher J,Kuster E,Bergstedt U,et al.Protein kinasedependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in Gram-positive bacteria[J].Mol Microbiol,1995,15:1049-1053.
[15]Nessler S,Fieulaine S,Poncet S,et al.HPr kinase/phosphorylase,the sensor enzyme of catabolite repression in Gram-positive bacteria:structural aspects of the enzyme and the complex with its protein substrate[J].J Bacteriol,2003,185:4003-4010.
[16]Irmler A,Forchhammer K.A PP2C-type phosphatase dephosphorylates the PII signaling protein in the Cyanobacterium synechocystis PCC 6803[J].Proc Natl Acad Sci U S A,2001,98:12978-12983.
[17]Gaidenko T A,Kim T J,Price C W.The PrpC serine-threonine phosphatase and PrkC kinase have opposing physiological roles in stationary-phase Bacillus subtilis cells[J].J Bacteriol,2002,184:6109-6114.
[18]Merzbacher M,Detsch C,Hillen W,et al.Mycoplasma pneumoniae HPr kinase/phosphorylase[J].Eur J Biochem,2004,271:367-374.
