HIV辅助受体CCR5常见小分子抑制剂抑制效果的综合评价*
2012-05-10焦诗卉
焦诗卉,何 淼
(中山大学生命科学学院,广东 广州 510275)
CC趋化因子受体5(CC chemokine receptor 5,CCR5)与大多数趋化因子受体一样,属于G蛋白偶联受体,介导的信号传导通路为Gq途径;它主要由以下几个部分组成:胞外N末端,3个胞外环,7个跨膜区域和胞内C末端[1]。CCR5是细胞膜上单核-巨噬细胞亲和性的HIV-1辅助受体,也是β趋化因子MIP-1α、MIP-1β、RANTES等的天然受体。
艾滋病(Acquired immunodeficiency syndrome,AIDS)是由人类免疫缺陷病毒(Human immunodeficiency virus,HIV)感染引起的。在HIV 的感染过程(图1)中,HIV与靶细胞融合,主要由包被糖蛋白gp120 和跨膜亚基gp41介导。gp120 与靶细胞上的CD4 分子和辅助受体(CCR5 或CXCR4)先后结合,导致gp41的构型发生改变,形成6 股α-螺旋束核心结构,将病毒包膜与靶细胞膜拉近,并发生融合,完成病毒进入宿主细胞的感染过程。因此,如果能够有效抑制CCR5的作用,就能抑制HIV进入靶细胞,从而有效预防或治疗HIV的感染。
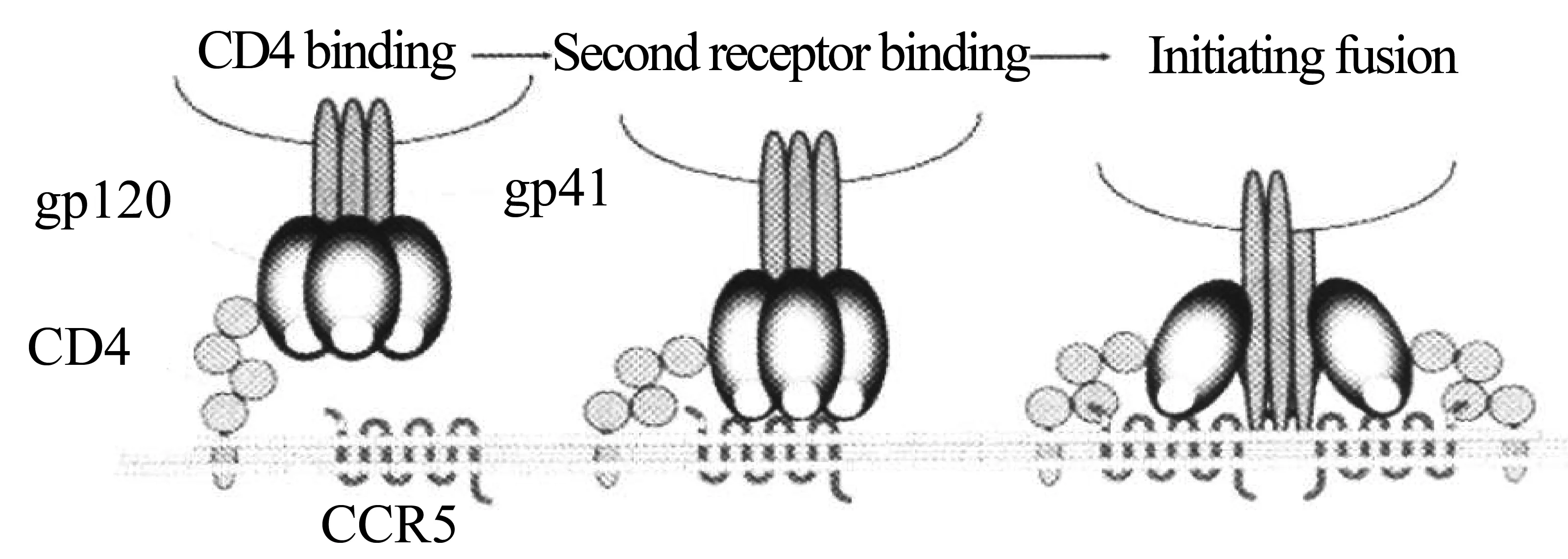
图1 HIV与细胞相互作用示意[2]
抑制剂的关键作用是抑制CCR5蛋白与gp120结合,从而有效阻碍HIV病毒进入靶细胞。1999年,自从日本Takeda公司成功开发出第一种CCR5小分子拮抗剂—TAK-779以来,针对CCR5的小分子抑制剂研究进展迅猛,相继出现了多种TAK-779的衍生物;例如,Yao Liu(2008)等人合成了TAK-779酮衍生物,证明其能有效抑制HIV-1。2007年,由辉瑞公司研发的CCR5趋化因子受体拮抗剂马拉韦罗(Maraviroc)成为了第一个被批准上市的CCR5拮抗剂药物。随后,SCH-C (SCH 351125)及其衍生物SCH 350634, SCH 350581,SCH 417690(Vicriviroc)也相继开发并应用于临床[3]。我国学者针对CCR5的小分子抑制剂进行的综述中,将目前开发并进入临床研究的小分子抑制剂分为7类:天然小分子类、苯并环庚烯类、哌啶类、螺环二酮哌啶类、托品烷类、4-哌啶-1-丁胺类和吡咯烷类[4]。
目前,有关CCR5常见抑制剂抑制效果的比较研究尚未见报道。本文通过构建CCR5常见抑制剂的三维结构,系统模拟各种抑制剂与CCR5蛋白分子的对接模式。通过比较各抑制剂对接模式的优劣,评估抑制剂的抑制效果;并尝试寻找CCR5蛋白的优化对接靶点,筛选CCR5的候选最优抑制剂,为相关理论研究和药物设计提供参考。
1 数据与方法
1.1 数据来源
CCR5蛋白三维结构信息下载自NCBI(http:∥www.ncbi.nlm.nih.gov/)的蛋白数据库。
CCR5小分子抑制剂的基本化学结构和基本分类方法参照刘瑶等(2007)的工作[4]。本文研究对象为CCR5小分子7类的29种常见抑制剂,主要包括:天然小分子类(抑制剂1-6)、苯并环庚烯类(抑制剂7-8)、哌啶类(抑制剂9-13)、螺环二酮哌啶类(抑制剂14-16)、托品烷类(抑制剂17)、4-哌啶-1-丁胺类(抑制剂18-23)和吡咯烷类(抑制剂24-29)。
1.2 数据处理与模拟分析
1.2.1 构建抑制剂的三维结构 采用Accelrys公司的Material Studio 5.5(MS)软件的Materials Visualizer模块,构建抑制剂的三维结构,按照药效团分类输出图形。
1.2.2 模拟小分子抑制剂与CCR5蛋白分子对接效果 使用Discovery Studio 2.5(DS)软件的LibDock模块模拟小分子抑制剂与CCR5蛋白分子的对接效果,根据抑制剂与靶点对接时的对接模式和亲和力进行综合打分。参数设置如下:Numbers of Hotspots为0.25;Docking Preference为User Specified; Conformation Method为BEST;Parallel Processing为True。
1.3 小分子抑制剂抑制作用评估
在分子对接的模拟过程,可以得到分子对接绝对自由能(Absolute Free Energy,记为AFE)、对接姿态数(Conf Number,记为CN)、相对自由能(Relative Free Energy,记为RFE),以及LibDock综合得分(LibDock Score,记为LDS)等参数值。其中,绝对自由能和相对自由能代表了该抑制剂与位点对接所需的能量,通常绝对自由能和相对自由能越低,表明该对接位点的对接越紧密,即抑制剂对该位点的抑制作用越强。对接姿态数越大,表明抑制剂与该位点作用的可能性越大。LibDock综合得分是LibDock模块综合绝对自由能、相对自由能和对接姿态数后,对该对接姿态的综合性打分;LibDock综合得分越高,抑制剂对该位点的抑制作用越强。最后,利用Excel将综合得分均值排序,分析和评估抑制剂与CCR5靶点相互作用效果。
2 结果与分析
2.1 构建小分子抑制剂的三维结构
基于MS软件,我们分别构建了7类抑制剂的三维结构。初步比较发现,各类抑制剂之间均具有部分相似的结构,该结构可能是抑制剂的药效团,即与CCR5结合的部位。
2.2 小分子抑制剂对接位点与评估
研究证实,小分子抑制剂与CCR5蛋白选择的结合位点主要在胞外部分[12-13]。同时,兼顾考虑到CCR5蛋白胞外N末端的结构特点,在CCR5蛋白潜在的13个可能结合位点中,我们缩小研究范围,主要关注site2,site4,site6和site8等4个结合位点(图3)。
2.2.1 CCR5的4个可能结合位点与小分子抑制剂的对接结果 综合各抑制剂对site2,4,6,8的LibDock综合得分, 以及各抑制剂与该4个位点结合的可能姿态数得到表1, 即各抑制剂与候选位点的LibDock综合得分均值以及可能结合姿态数占总姿态数的百分比。
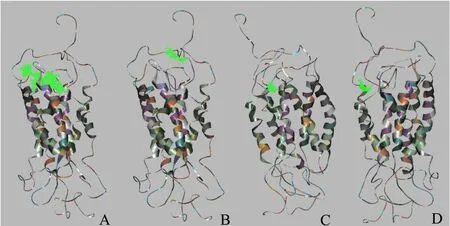
图3 CCR5蛋白的4个可能结合位点

表1 CCR5可能的结合位点与小分子抑制剂结合综合得分
在表1中,初步比较4个位点的LibDock综合得分,可知Site 4的综合得分最高,表明小分子抑制剂在该位置与CCR5蛋白结合最紧密。
分别统计4个位点所有可能的对接姿态数目占总的可能结合姿态数目的百分比(Docking Attitude Percent,记为DAP),发现在site 4,对接姿态百分比达到了96.05%。因此,推断CCR5的site 4是小分子抑制剂的主要作用位点,该位点也可能是抑制效果最优的位点。
2.2.2 小分子抑制剂的抑制作用分析与评估 系统计算各类小分子抑制剂与CCR5的site4结合的主要参数,包括AFE、RFE、GP和LDS的均值。具体计算结果见表2。

表2 各类小分子抑制剂与CCR5的site4结合的相关参数
依照LibDock综合得分排序,抑制剂5、8、12、14、17、21和27在各自类别内综合得分最高。因此,这7种抑制剂对CCR5蛋白的抑制作用可能相对较强。比较对接姿态百分比与LibDock综合得分2个参数值,发现LibDock综合得分的高低与对接姿态百分比的大小并无直接联系。同时,考虑对接的相对自由能和绝对自由能,进一步发现,该3个数值共同影响了LibDock的综合得分。
在天然小分子化合物类别中,通过比较LibDock综合得分,初步得到该类别抑制剂的排序,即:抑制剂5>抑制剂1>抑制剂4>抑制剂6>抑制剂3>抑制剂2。Yoganathan等(2003)的实验结论发现,甲氧基二氢暗褐菌素(抑制剂1)、暗褐菌素(抑制剂2)和Fuscinarin(抑制剂3)对CCR5蛋白抑制的 IC50值分别为154、21和80 μmol/L[5],支持了我们的结论。该IC50值可以表示化合物对CCR5蛋白的抑制作用, 因此该值越大,表明对CCR5蛋白的抑制作用越强。
另一部分学者研究抑制剂对[125I]-RANTE与表达CCR5的中国仓鼠细胞(CHO) IC50值,该值表明了化合物对RANTE的抑制作用,因此数值越大,对RANTE的抑制作用越强,并对CCR5的抑制作用越弱。根据他们的研究,研究发现Sch351125(抑制剂9)对[125I]-RANTE与表达CCR5的中国仓鼠细胞(CHO)的IC50值为9.0 nmol/L[6];Sch417690(抑制剂12)的抗病毒活性要比Sch351125(抑制剂9)高近10倍,其对R5型HIV-1的IC50值为0.1~3 nmol/L[7]。Wood A等[8]研发了进入临床Ⅲ期的小分子抑制剂UK-427857(抑制17),可以有效阻断 [125I]-RANTE与CCR5的结合,其IC50值为7.18 nmol/L。以上实验结果支持了计算得到抑制剂的抑制效果排序的结论,即:抑制剂12>抑制剂17>抑制剂9。
此外,默克公司研制的1,3,4-三取代吡咯烷化合物(抑制剂24),其抗CCR5的IC50值为26 nmol/L。根据该类化合物改进获得的抑制剂28和29,其IC50值分别为0.13和2.5 nmol/L[9]。同样支持了关于吡咯烷类化合物抑制效果排序的计算推断,即:抑制剂28>抑制剂29>抑制剂24。
2.2.3 候选抑制剂与site 4对接效果模拟 基于Discovery Studio软件,可以获得抑制剂与CCR5蛋白对接的结果,放大对接位点,去掉未与位点结合的分子,分析与位点结合的分子个数,以及所占的位点面积(图4)。
初步比较发现,抑制剂8(图4B)与site4的结合区域明显大于其它抑制剂。按照结合区域的大小排序,依次为抑制剂14(图4D)、抑制剂27(图4G)、抑制剂21(图4F)、抑制剂12(图4C)、抑制剂5(图4A)和抑制剂17(图4E)。
但是,基于LibDock综合得分得出的抑制剂效果排序为:抑制剂27>抑制剂8>抑制剂12>抑制剂14>抑制剂21>抑制剂5>抑制剂17。两种方式获得排序不完全一致,表明结合区域的大小与抑制剂的抑制效果不完全具有正相关关系。可能是结合部位的分子类型或分子结构会影响到LibDock综合得分。

图4 抑制剂5、8、12、14、17、21和27分别与site4结合姿态放大图
3 讨 论
CCR5是目前治疗HIV-1的新型靶点,有效抑制CCR5便能够有效控制HIV-1病毒感染细胞。目前,国际上针对CCR5抑制剂的研究倍受关注。本文利用Accelrys公司的软件Discovery Studio的LibDock模块成功模拟了小分子抑制剂与CCR5蛋白的对接,计算推测了抑制剂的抑制效果排序,模拟了对接效果,为进一步药物开发和临床使用提供了参考。
计算分析得到了抑制剂的抑制效果排序,即:抑制剂27>抑制剂8>抑制剂12>抑制剂14>抑制剂21>抑制剂5>抑制剂17;并且,初步发现CCR5蛋白与小分子抑制剂结合的位置可能主要在第二个胞外环。因此,未来的小分子抑制剂的开发应着眼于CCR5的第二个胞外环,对哌啶类的抑制剂27进行修饰可能会发现具有更强抑制效果的抑制剂。
然而,相关实验检测吡啶类生物碱anibamine的三氟醋酸盐(抑制剂6),蛇胞菌素c(抑制剂4) 和19,20-环氧松胞菌素Q(抑制剂5)对CCR5的抑制效果,获得的IC50值分别为1、40和60 μmol/L[10]。实验结果与计算推测结论:抑制剂4>抑制剂5>抑制剂6,并不完全符合。可能的原因是,软件设计中未考虑到蛋白与小分子抑制剂在对接时可能发生构象的改变。
另外,TAK-220 (抑制剂23)具有很好的CCR5拮抗作用,IC50值3.5 μmol/L;它可以与CCR5上的第4、5、6跨膜区结合[11],这与实验的结论有所不同。可能的原因在于,本文分析了与大多数抑制剂结合的CCR5第二个胞外环,而TAK-220是一个较特殊的抑制剂,与大多数小分子抑制剂结构差异较大,这可能是导致抑制剂23抑制作用较低的一个重要原因。
虽然小分子抑制剂对CCR5具有很强的抑制作用,但是,它们易造成抗性的产生,导致拮抗剂的功能下降或丧失。通常小分子抑制剂只能作用于CCR5的单个位点,而多肽抑制剂则能够与CCR5蛋白相互作用,从而结合多个位点。因此,多肽抑制剂具有更高的临床意义,这也可能是未来CCR5拮抗剂开发的方向。
参考文献:
[1] DEL CORNO M,LIU QH,SCHOLS D,et al. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling [J]. Blood,2001,98(10):2909-2916.
[2] 韩燕星,蒋建东. CCR5:HIV-1药物的新靶点[J]. 中国医学科学院学报,2003,25(5):635-639.
[3] CECILE L.T,FRANCOISE G,C HRISTOPHER K,et al. Anti-human immunodeficiency virus interactions of SCH-C(SCH 351125), a CCR5 antagonist with other antiretroviral agentsinvitro[J]. Antimicrobial Agents and Chemotherapy,2002,46(5):1336-1339.
[4] 刘瑶,苏靖,李松. CCR5小分子拮抗剂类抗艾滋病药物研究进展[J]. 国外医学药学分册,2007,1(34):7-11.
[5] YOGANATHAN K,ROSSANT C,NG S,et al. 10- Methoxydihydrofuscin,fuscinarin,and fuscin,novel antagonists of the human CCR5 receptor from Oidiodendron griseum [J].J Nat Prod,2003,66(8):1116-1117.
[6] PALANI A,SHAPIRO S,CLADER J W,et al.Discovery of 4-[(z)-(4-bromophenyl)- (ethoxyimino)methyl]-1'-[(2,4-dimethyl-3-pydinyl)carbonyl]-4'-methyl-1,4'-bipiperidine N-oxide(SCH351125):an orally bioavailable human CCR5 antagonist for the treatment of HIV infection [J].J Med Chem,2001,44(21):3339-3342.
[7] STRIZKI JM,TREMBLAY C,XU S,et al.Discovery and charaeterization of vicriviroc(SCH 417690),a CCR5 antagonist with potent activity against human immunodeficiency virus type 1 [J].Antimicrob Agents Chemother,2005,49(12):4911-4919.
[8] WOOD A,AMLOUR D.The discovery of the CCR5 receptor antagonist,UK-427,857,a new agent for the treatment of HIV infection and AIDS [J].Prog Med Chem,2005,43:239-271.
[9] LYNCH C L,GENTRY A L,HALE J J,et al.CCR5 antagonists:bicyclic isoxazolidines as conformationally constrained N-1-substituted pyrolidines [J]. Bioorg Med Chem Lett,2002,12(4):677-679.
[10] JAYASURIYA H,HERATH K B,ONDEYKA J G,et al. Isolation and structure of antagonists of chemokine receptor(CCR5) [J]. J Nat Prod,2004,67(6):1036-1038.
[11] MASAO N,KATSUNORI T,TOSHIYA N,et al. Analysis of binding sites for the new small-molecule CCR5 antagonist TAK-220 on human CCR5 [J]. Antimicrobial Agents and Chemotherapy,2005,49(11):4708-4715.
