硫氰酸盐比色测定钼的方法研究
2012-04-27徐志昌张萍
徐志昌,张萍
(清华大学核能与新能源技术研究院,北京102201)
0 引言
在工艺试验研究中,工艺人员对硫氰酸盐比色测定结果的离散型,往往扑朔迷离,不知所措,特别是,钨-钼共存的样品,测试结果的不统一,是屡见不鲜的事情。这种现象是怎么产生的呢?
笔者认为它是由下列3项因素造成的,即:(1)还原与络合的不稳定性;(2)比色设备落后;(3)钨-钼比色的相互干扰性。采用硫氰酸盐比色测定钼是应用较广的分析方法[1-6]。该方法与重量法比较,具有简便、快捷等优点。比色测定钼的机理,首先是添加还原剂将Mo(Ⅵ)还原成Mo(Ⅴ),然后,再与硫氰酸盐反应并转变为组成与结构确定的配合物,最后作吸光度的测定,或者通过萃取转型再作吸光度测定。
本文在硫酸介质中使用硫酸铜催化,硫脲为还原剂,使Mo(Ⅵ)还原为Mo(Ⅴ),并与硫氰酸盐组成配合物,以作吸光度测定。其最大吸收波长是根据吸收光谱曲线来确定的;浓度的测定是依据标准曲线来确定的。结果表明,硫氰酸盐与Mo(Ⅴ)生成的络合物有最大吸收波长位置为457.90 nm;根据标准曲线确定的线性方程式:A=0.122 96C+ 0.003 39。其相关系数=1.000 0。此方法的分析结果稳定,重现性好,操作简便、迅速。
比色测定文献表明,比色测定的发展趋势,不仅包括还原剂的改进,而且包括光谱仪器的改进。通常,W(Ⅵ)的还原较难,Mo(Ⅵ)还原较易。前者采用双无机还原剂,如 SnCl2-TiCL3[2]、SnCl2-NaH2PO2[8]等;后者,则采用有机还原剂,如抗坏血酸和硫脲等,同时添加阳离子作催化剂[3-5]。
作为Mo(Ⅵ)的还原剂,可以有不同的选择,其中包括有机还原剂:硫脲和抗坏血酸等有机化合物作为Mo(Ⅵ)的还原剂,研究和使用较多的是抗坏血酸[3,5,6]和硫脲等。有机还原剂(添加催化剂),与无机还原剂相比较,其灵敏度与和稳定性都有所提高。
和直接比色法比较,萃取法比色法具有很多优点,其中包括提高了选择性、灵敏度与稳定性,可以消除三氯化钛紫色对比色的干扰,但是,由于分析程序复杂,通常只在钼浓度很低的情况下需要使用。萃取溶剂通常是由30%乙酸乙酯与苯溶剂组成的溶液,例如,硫氰酸盐-乙酸乙酯比色测定钢中钼含量。
追根穷源,比色法的理论依据是Lambert-beer定律,即单色光透过吸光体,其光强度的降低同入射光波长、吸收介质的厚度及光路中微粒的数目成正比。其数学表达式是:

式中I0是入射光强度;I是透射光强度;K0是吸光系数;K1是直线截距;b是比色槽厚度;c是被测溶液的配位化合物分子浓度。
公式表明,配位分子对光的吸收不仅具有选择性,而且与配位分子的空间结构与入射光波长有关,因此,比色法测量的第一步是要建立吸收光谱曲线,以选择最佳波长,然后再选择配位化合物的形成与稳定条件,以建立标准曲线。
传统的比色测定过程是用作图法画出标准曲线,由于作图有较大的随意性,尤其在测量数据比较分散时,对同一吸光度数据,不同的分析者可以得出不同的结果,因此,这是一种粗略的数据处理方法。如果,同样的吸光度数据让计算机软件来处理,即采用最小二乘法将一组符合线性方程:y=a+bx关系的测量数据用计算方法求出最佳的a和b,那么,任何人做出的结果都是唯一的。
1 试验方法
1.1 比色分析试剂配制以及固体样品的化学分解
1.1.1 比色分析试剂的配制
CuSO4+H2SO4溶液:浓度分别是0.1%CuSO4+9 mol/L H2SO4;
KCNS:25%水溶液;
硫脲:5%的水溶液。
1.1.2 固体样品的化学分解
比色分析是以液体样品为前提的,因此,一切固体样品均需通过化学分解过程将其转化为液体样品。固体样品的化学分解对于被测定元素的灵敏度与准确性具有重要作用。表1是金属与矿物固体样品的化学分解方法与转化过程。由表1可见,固体样品的分解与转化经常采用酸法,而较少使用碱熔融法。
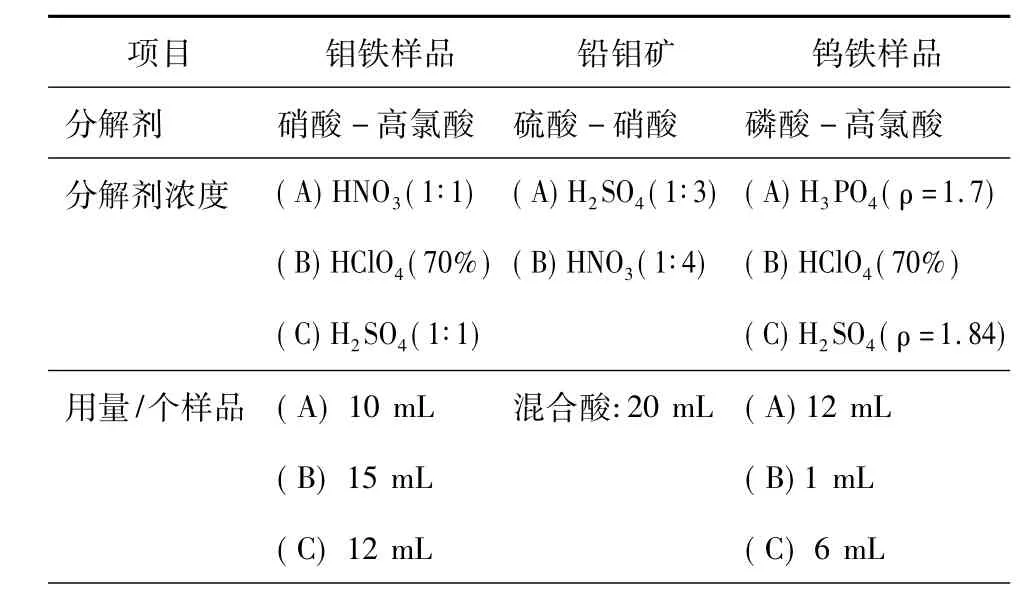
表1 固体样品的化学分解与转化过程
1.2 仪器与设备
SP752PC;采用紫外可见光光度计,连接计算机后,可实现双向通讯。该机具有自检、自校波长、光源自动切换、用计算机控制仪器等功能。可以通过测试标准样品的吸光度,输出标准样品的浓度,实现8个样品二阶线性拟合,从而获得标准曲线。它们可以通过存储、打印或调用曲线参数进行未知样品的测量。
在仪器的波长范围内,根据设定的波长范围,对样品进行光谱扫描,测试样品的吸光度、透射比随波长的变化曲线,分析样品的光谱特性,寻找样品的最大吸收峰位置,可对几组图谱进行叠加和混合四则运算,可打印和存储图谱。
1.3 分析方法与步骤
稀释倍数按照样品浓度高低来确定,低浓度溶液可以直接还原和发色;高浓度溶液则选择100~1 000倍来取样。以30 mL无离子水稀释样品,同时以自来水冷却到常温。然后,添加9 mL催化剂溶液以及10 mL硫脲溶液,然后,等待10 min后进行比色测定。
2 试验结果与讨论
2.1 分析流程
(1)取样与稀释:为了防止钨的水解,稀释前应加入适量碱溶液。通常,预先加入5 mL NaOH溶液。因为,制订标准曲线的钼浓度单位是mg/L,所以,对于1 mL溶液而言,通常是g/L的样品,需要稀释1 000倍,方可进行检测。
(2)添加催化剂:有机还原剂,如硫脲、抗坏血酸等对于W(Ⅵ)和Mo(Ⅵ)的还原,往往需要添加像Cu(Ⅱ)、Fe(Ⅲ)等离子作催化剂,以加速与稳定还原过程。因此,本实验添加9 mL的0.1%CuSO4,9 mol/LH2SO4溶液作还原催化剂。由于硫酸具有较大的稀释热,加入30 mL水后,迅速冷却至常温。

表2 有机还原剂的比较
(3)发色时间:随着温度变化而变化,温度越低,时间越长,反之越短。通常温度下,采用10 min的发色时间。
图1是硫氰酸盐比色测定钼的流程框图。由图1可见,液体样品的稀释是必要的步骤。通常,钨钼溶液的浓度是 g/L。而比色测定的浓度范围是mg/L,需要稀释1 000倍。浓度越高,稀释倍数越高;反之,浓度越低,稀释倍数越低。
试验表明,即使在一定酸度条件下,高价钼,Mo (Ⅵ)的还原,以及硫氰酸钼,Mo(Ⅴ)络合物的生成速度较慢,需要一定的还原时间来完成。

图1 硫氰酸盐比色测定钼的流程框图
2.2 还原剂和络合剂的比较
使高价钼Mo6+还原为低价Mo5+的试剂,经常被使用和研究的有,SnCl2、TiCl3、硫脲以及抗坏血酸等。它们的使用量和介质条件各不相同。表3列举了各自的参数与使用性能。
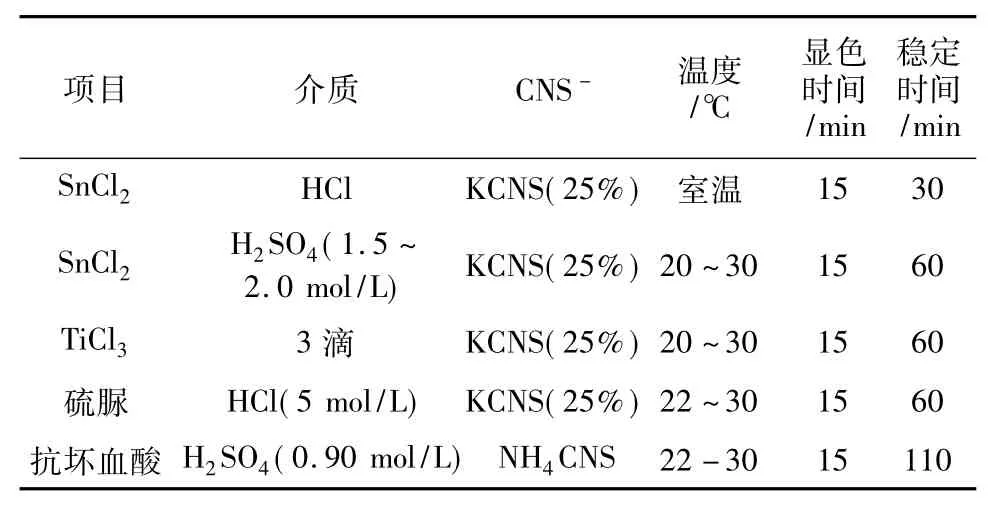
表3 Mo(Ⅵ)还原剂的使用与比较
文献[6]研究了钼铁产品中钼的硫氰酸盐比色测定方法。研究以高氯酸铁为抑制剂,抗坏血酸为还原剂的硫氰酸盐分光光度法,测定了高含量钼的方法。钼铁试样用硝酸和高氯酸溶解,硫酸冒烟,盐类溶解后在高氯酸铁的存在下,以抗坏血酸还原Mo(Ⅵ)为Mo(Ⅴ),Mo(Ⅴ)与硫氰酸铵生成橙色络合物,在460 nm波长处测定其吸光度。用本法测定了3个钼铁标样中钼,测定值与认定值相符,6次测定的相对标准偏差为0.29%~0.35%,分析时间只需50 min。而重量法的分析周期为10~12 h。
优化参数包括:5 mL抗坏血酸(5%);4 mLNH4CNS(30%);5 mL Fe(ClO4)3(1%);5 mL H2SO4(1∶1);22~30℃(15 min),冬季加温。其中,铁离子的作用被称为抑制剂,似乎不妥,应当改为催化剂。这是因为,其作用是加速还原与稳定发色。
文献[5]通过硝酸-硫酸混合溶液分解固体铅锌矿样品,然后采用抗坏血酸—硫氰酸盐分光光度法测定浸取液中钼含量,在选定的条件下。该方法的(Mo)检出限为0.076 3 μg/mL,(Mo)线性范围为0.4~6.768 μg/mL,加标回收率为94% ~99%,用此法测定铅锌矿中钼含量,结果满意。
溶液取量与配置浓度见表4。
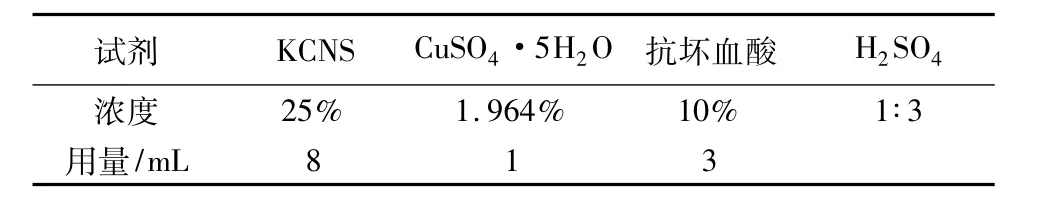
表4 标准溶液的浓度与取量
2.3 硫酸浓度的影响
硫氰酸钼络合物的吸光度也和酸度有关,图2是硫酸克当量浓度对络合物吸光度的依赖曲线。曲线表明,吸光度随着酸度而增加,超过2 g当量浓度后趋于稳定。

图2 吸光度对硫酸浓度的依赖曲线
2.4 硫氰酸钼的分子吸收光谱曲线
图3是硫氰酸钼的分子吸收光谱图。由图3可见,我们从可见光区,波长为600 nm开始扫描,被发现的第一个最大吸收峰是459 nm(吸光度=1.27),其后的许多峰谷,其中包括359.9 nm、302.0 nm、301.8 nm、300.5 nm、297.3 nm等,虽然吸光度很大,但选择性很差,不是特征峰。因此,确定波长459 nm是最合理的选择。

图3 硫氰酸钼分子吸收光谱图
2.5 硫氰酸钼分子吸收的标准曲线
对于受计算机控制的紫外可见光分光光度计而言,放弃了传统的手工方法,其精确度与准确性有很大提高。实现8个样品的一阶线性拟合做好的标准曲线以及线性方程,可以存储、打印、或调用曲线参数来进行未知样品的结果计算。图4是硫氰酸钼吸光度与浓度之间的关系曲线。方程式(1)是硫脲-硫酸铜催化还原钼所得线性方程。
Abs=0.00339+0.12296C (1) C=[Abs-0.00339]/0.1229=8.137Abs-0.0276 (2)
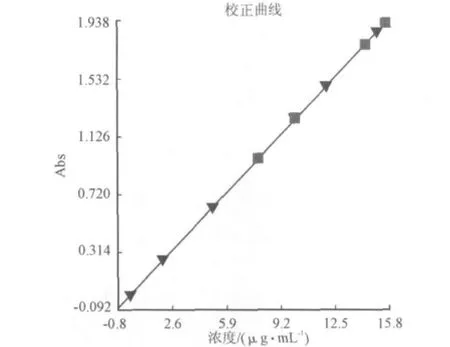
图4 硫氰酸钼吸光度与浓度之间的关系曲线
3 讨论
3.1 分析仪器
传统的比色测定过程是用作图法画出标准曲线,由于作图有较大的随意性,尤其在测量数据比较分散时,对同一吸光度数据,不同的分析者可以得出不同的结果,因此,这是一种粗略的数据处理方法。如果,同样的吸光度数据让计算机软件来处理,即采用最小二乘法将一组符合线性方程:y=a+bx,关系的测量结果用计算数据处理方法,来求出最佳的a和b,那么,任何人做出的结果都具有唯一性。
通常,在工厂企业里,大多选择价格便宜的分光光度计,即721型或722型分光光度计,它们只有一种单色光源,波长范围是可见光区(400~1 000 nm),其;步长选择受到人为的限制,吸光度为纵坐标,波长为横坐标取得的光谱曲线以及最大吸收峰位置,亦有随意性。
近几年来,随着我国光谱仪器以及计算机应用于软件工程的发展,受计算机控制的紫外可见光光谱也得到了快速发展。例如,上海光谱仪器公司的产品SP752PC、SP754PC等。连接计算机后,可实现双向通讯。该机具有自检、自校波长、光源自动切换、用计算机控制仪器等功能。可以通过测试标准样品的吸光度,输入标准样品的浓度,实现8个样品一阶线性拟合,从而获得标准曲线。它们可以通过存储、打印或调用曲线参数进行未知样品的测量。
3.2 还原剂
Mo(Ⅵ)的还原比W(Ⅵ)来得容易,因此,选择有机还原剂的灵敏度和稳定性更好。本文采用硫脲与硫酸铜催化的还原方法使Mo(Ⅵ)还原为Mo (Ⅴ),并与硫氰酸盐生成橙红色络合物,借以比色测定。文献[8]探讨了抗坏血酸还原的硫氰酸盐比色测定钼的方法。
对于低含量钼样品(0.1%),国标(GB/T-223.27-94)采用乙酸丁酯萃取比色法测定钼。总之,对于钼而言,其还原剂采用有机还原剂,其中包括硫脲和抗坏血酸等,其灵敏度与稳定性都是满意的,深受研究者与厂矿企业的欢迎。
3.3 萃取比色
由于钨和钼具有相似的化学行为,因此,它们彼此的比色测定将相互干扰。消除干扰的最佳方法是通过萃取方法分离它们。通常,它们与硫氰酸盐络合物的萃取能力存在明显差异。因此,通过萃取优先萃取钨是必要的。在动力学方面的差异是硫氰酸钨的萃取速度较快,即利用快速萃取方法使得钨和钼获得分离。图5是N-235对钨-钼萃取动力学曲线。曲线表明,N-235对钨的萃取速度明显地高于钼。这就是萃取比色测定钨的理论依据。
4 结论
上述结果表明,比色分析结果的准确性在工艺研究中占有举足轻重的地位。如果分析结果不准确,那么工艺研究将举步维艰。工艺研究人员应当关心、重视分析方法。二者应协调发展,共赢前进。下列结论是显而易见的:
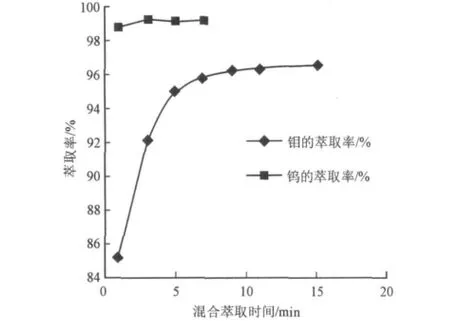
图5 N-235对钨-钼萃取动力学曲线
(1)比色测量结果的准确性首先取决于还原与络合的稳定性:硫氰酸钼(Ⅴ)的形成,不仅要以还原为前提,而且要以络合物存在的酸度、时间等为条件。Mo(Ⅵ)的以还原性,仅仅需要较弱的有机还原剂,例如,抗坏血酸和硫脲等,在无机离子(Cu(Ⅱ)、Fe(Ⅲ)等)催化下,稳定地完成。
(2)其次,比色测定的准确性取决于比色计的水平。传统的可见光分光光度计,其光谱曲线与标准曲线的正确性,远不及先进的紫外可见光光度计。后者不仅可以获得精确的光谱曲线,而且,可以获得精确的标准曲线,从而避免了传统比色分析设备的人为误差。根据吸收光谱曲线来选择最大吸收波长;根据标准曲线来求得线性回归方程,再根据回归方程来确定未知样品的含量。在最大吸收波长下获得回归系数为1的标准曲线。
(3)最后,硫氰酸盐比色测定的正确性还取决于消除相互干扰的手段。硫氰酸W(Ⅴ)与硫氰酸Mo(Ⅴ)的最大吸收峰,比较接近(分别是405 nm、460 nm),比色干扰在所难免。特别是它们在含量比较接近的情况下,相互干扰,在所难免。利用萃取分离比色是分析工作者的最佳选择。
[1]张必成,陈沛智.仪器分析[M].武汉:湖北出版社,1997,66-84.
[2]杜治坤,杨素卿.硫氰酸盐萃取比色法测定三氧化钼、钼精矿中微量钨[J].湖南冶金,1976,(4).
[3]徐伯洪,鲁雁飞,肖宏瑞.用抗坏血酸作还原剂测定空气中钼[J].卫生研究,1980,(4).
[4]GB/T223.27-1994,钢铁及合金化学分析方法[S].硫氰酸盐-乙酸乙酯萃取分光光度法测定钼量.
[5]陈忠书,金绍祥.硫氰酸盐光度法快速测定铅锌矿中钼[J].矿产与地质,2007,(3):381-382.
[6]汤成兰,王兆存,刘景华.硫氰酸盐分光光度法快速测定钼铁中钼[J].冶金分析,2007,(11):78-79.
[7]GB/T14352.1-1993,钨矿石、钼矿石化学分析方法[S].硫氰酸盐光度法测定钨量.
[8]张明德.抗坏血酸-硫氰酸盐光度法测定钼的探讨[J].特钢技术,2003,(3):25-29.
