PAN/氧化石墨烯纳米复合材料的热性能研究
2012-03-25陈宜波陈友汜欧阳琴
陈宜波,陈友汜,欧阳琴,皇 静,严 庆
(中国科学院宁波材料技术与工程研究所碳纤维制备技术与工程国家工程实验室,浙江宁波315201)
聚丙烯腈(PAN)纤维是制备高性能碳纤维的一种重要前驱体。在热稳定化过程中PAN通过氰基基团的环化反应以及分子链间的交联氧化脱氢反应等放热反应形成耐热稳定的梯形结构[1]。目前,PAN基碳纤维的强度与理论值相距较大,H.G.Chae等[2]曾预言新一代 PAN 基碳纤维应该是由PAN与碳纳米管(CNTs)等构成的复合纤维。H.G.Chae等还专门研究了PAN/CNTs复合纤维的热稳定化过程和力学性能,结果显示CNTs的引入抑制了PAN的环化反应,同时也显著提高了PAN纤维的拉伸强度和弹性模量[3-4]。石墨烯作为一种新型的纳米碳材料,它是由单层碳原子紧密堆积而成并呈现出二维蜂窝状的晶格结构[5]。与CNTs相比,石墨烯不仅具有更优异的导热导电性,而且可由天然石墨经过简单工艺制备得到,不含金属催化剂,成本低廉。另外,石墨烯的前驱体氧化石墨烯(GO),表面含有丰富的含氧官能团,能提高其溶液加工性能,增强与极性聚合物的相互作用[6]。基于纳米碳材料与PAN基体具有良好相容性的优势,作者采用均相溶液聚合的方法制备了共聚PAN体系下PAN/GO的纳米复合物,研究了GO对自由基聚合过程的影响,并进一步探讨了其对 PAN热性能的影响,为开发新一代PAN基碳纤维材料打下基础。
1 实验
1.1 原料
天然石墨:平均粒径2 μm,青岛久益石墨有限公司产;丙烯腈:浙江台州中海医药化工有限公司产,减压蒸馏后冷藏备用;偶氮二异丁腈(AIBN):上海四赫维化工有限公司产;衣康酸(IA):阿拉丁化学试剂有限公司产;高锰酸钾、双氧水、二甲基亚砜(DMSO):均为分析纯,上海国药化学试剂有限公司产。
1.2 仪器
Nicolet 6700傅里叶变换红外光谱仪:美国Thermo Fisher Scientific公司制;Lambda 950型紫外可见近红外分光光度计:美国Perkin Elmer公司制;RV DV-Ⅱ +PRO型旋转黏度计:美国Brookfield公司制;S4800型扫描电子显微镜(SEM):日本日立公司制;Tecnai F20型透视电子显微镜(TEM):美国FEI公司制;STA 449 F3型差示扫描量热-热失重(DSC-TG)热分析仪:德国耐驰公司制。
1.3 GO 的制备
采用改进的Hummers方法[7]制备GO。称取1.5 g石墨到烧杯中,室温下加入60 mL质量分数为80%的浓硫酸,磁力搅拌15 min。升温至40℃,缓慢加入9 g高锰酸钾,恒温搅拌加热2 h。紧接着升温至85℃,加入75 mL去离子水,继续搅拌加热约30 min,稀释的混合液变成黄褐色后,再次加入300 mL去离子水,并在此之后逐滴加入20 mL质量分数为30%的双氧水,借此中和未反应的高锰酸根,溶液最终变成金黄色悬浮液。将悬浮液静置过夜,待其沉降完全,倒去上层清液,继续用去离子水稀释并搅拌不少于2 h,静置沉降后继续重复水洗操作。水洗后期悬浮液无法完全沉降,改自然沉降为离心沉降,水洗周期不少于15次。最后的离心沉降物即为高浓度GO浓缩液,冷冻干燥24 h,得到蓬松的试样,备用。气体吸附性能测试表明该GO的比表面积达到312.5 m2/g,与文献[8]报道值相近。
1.4 PAN/GO纳米复合材料的制备
采用以AIBN为引发剂的传统均相自由基聚合,AIBN占单体质量的0.8%,在双层玻璃聚合釜中进行反应。复合体系聚合前,称取1 g GO(占单体总质量的1%),将其分散在400 mL DMSO中,采用细胞粉碎机超声处理20 min得到均一稳定的分散液,将引发剂和IA(占丙烯腈质量的1%)搅拌溶解在部分GO分散液后连同100 g丙烯腈依次加入到聚合釜中。在40℃和氮气保护条件下以50 r/min的搅拌速度搅拌30 min,再将温度升高到60℃,聚合24 h。研究聚合过程时,从釜底阀门定时取样。
1.5 测试
红外光谱:使用红外光谱仪测试试样的吸收光谱,用KBr粉末压片法进行制样。
紫外吸收光谱:使用紫外可见近红外分光光度计测试试样的紫外吸收。
黏度:使用旋转黏度计在60℃条件下测试聚合液黏度。
形貌结构:使用SEM和TEM观察试样,薄膜试样经环氧树脂包埋并用超薄切片制样后观察获得其TEM照片。
热性能:使用DSC-TG热分析仪测试试样的热性能,升温速率为10℃/min,在空气气氛下从室温扫描至900℃。
转化率:将约2 g聚合液压平压薄后水洗并水煮30 min,烘干后称量,根据水洗干燥前后的质量变化和投料固含量计算转化率。薄膜试样的制备:将聚合液用玻璃棒在光滑玻璃板上均匀刮涂一层厚度小于2 mm的薄层,在80℃下真空干燥24 h除去溶剂。
2 结果与讨论
2.1 GO对自由基聚合反应的影响
从图1可以看出,在同一时间点上,与空白试样PAN相比,复合体系聚合液的黏度和单体转化率均较低,说明GO在一定程度上阻碍了自由基聚合。这种阻聚作用在聚合前期表现得比较明显,越到后期越弱化。聚合至第13 h时,PAN/GO聚合液黏度为 30.9 Pa·s,其单体转化率为81.5%。与空白试样相比,复合体系聚合至第13 h时,聚合液的黏度和单体转化率分别降低了1.3%和 2.9%。

图1 聚合液黏度和单体转化率随聚合时间的变化
2.2 红外光谱和紫外光谱分析
从图2可以看出,GO谱图上3 423,1 726 cm-1和1 058~1 403 cm-1处的吸收峰分别对应GO上的—OH,—C=O 和—C—O 的伸缩振动[9]。PAN 在 2 242 cm-1处的强吸收峰为氰基基团的特征吸收峰,1 716 cm-1为共聚单体IA的羧酸基团的特征峰。利用羧酸基团的吸收强度(I1716)与氰基基团的吸收强度(I2242)的比值(I1716/I2242)可定量计算PAN大分子链上共聚单体的相对含量[10]。经过计算得知,PAN的I1716/I2242值为0.25,而 PAN/GO对应的值为0.30,这是因为GO的引入增加了复合体系羧酸基团的相对含量。
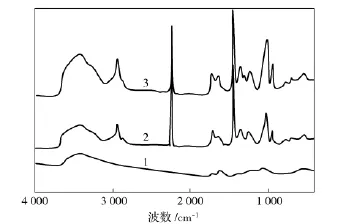
图2 试样的红外光谱Fig.2 FTIR spectra of samples
从图3可看出,GO的吸收峰值在299 nm,这代表了GO上碳碳共轭结构的特征吸收[11]。PAN除了在263 nm出现氰基基团上π键的特征吸收外,在350~400 nm波段还出现了较弱的共聚单体羧酸基团的特征吸收。在PAN/GO复合物中,氰基基团的特征吸收峰红移到271 nm,表明强极性的PAN与GO存在较强的π-π相互作用。
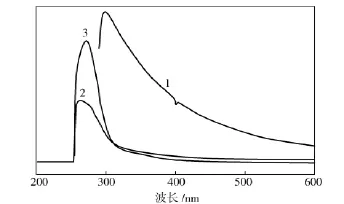
图3 试样的紫外光谱Fig.3 UV-Vis spectra of samples
2.3 形貌结构
由图4可看出:GO试样经冷冻干燥后呈现出相互堆叠的片层形状,这些片层由透明的超薄GO组成,其间有部分相互接触,只需在溶液中简单超声可得到孤立的GO;同时,还观察到GO表面充满波浪状条纹和褶皱,根据报道,完美的超薄二维石墨烯单晶是热力学不稳定的,因此需要在其表面形成条纹和褶皱来保持形态稳定[12]。在PAN/GO复合膜的断面形貌SEM图(图4b)中可看到GO片层均匀分散在PAN基体中,并与基体接触紧密,说明两者具有很好的相容性。从图4c可看到,GO由彼此平行排列的单层石墨烯组成,其总厚度为3~4 nm;表明氧化石墨远程有序的结构已不复存在[13]。图4d显示了镶嵌在 PAN基体中的GO以单层(1 nm)形式存在,表明经聚合前的超声处理后通过原位插层聚合的方法可得到GO分散均匀的PAN/GO纳米复合材料。

图4 试样的SEM和TEM照片Fig.4 SEM and TEM images of samples
2.4 GO对PAN热稳定化过程的影响
从图5可见,PAN在空气气氛中的DSC曲线显示了其热稳定化的放热过程。PAN的DSC曲线出现2个放热峰,其中处于较低温度的放热峰归属于PAN大分子链上氰基基团的环化反应,而处于较高温度的第二个放热峰归属于PAN大分子链间的交联氧化脱氢反应[1]。通过对比观察可以看到,在PAN/GO复合物中GO的引入使环化反应放热峰移向高温,说明GO在一定程度上抑制了环化反应的进行,可能是因为处于GO附近的PAN与GO之间存在较强的π-π相互作用,这种强相互作用使得部分环化反应需要在更高的能量条件下才能引发进行。另外,在PAN/GO的DSC曲线中,环化反应放热峰在低温段出现了一个新的肩峰,这验证了复合物中的氰基基团因距离GO片层位置的远近不同而存在两种状态,致使环化反应分步进行。从图5还可以看出,GO对PAN的氧化脱氢反应的影响并不明显。
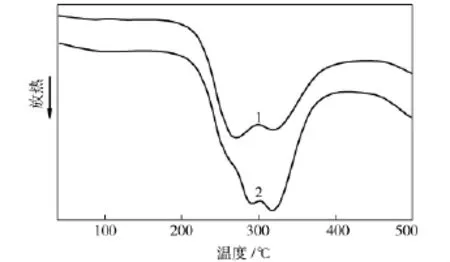
图5 试样的DSC曲线Fig.5 DSC curves of samples
3 结论
a.采用原位聚合的方法制备了PAN/GO复合材料,GO在聚合前期对自由基聚合起到一定的阻聚作用,到聚合后期这种阻聚作用有所弱化。
b.采用原位聚合的方法可以使GO以单层的形式分散在PAN基体中。
c.PAN与GO存在较强的π-π相互作用,这种相互作用抑制了PAN在热稳定化过程中氰基基团的环化反应。
[1] Bahrami S H,Bajaj P,Sen K.Thermal behavior of acrylonitrile carboxylic acid copolymers[J].J Appl Polym Sci,2003,88(3):685-698.
[2] Chae H G,Kumar S.Making strong fibers[J].Science,2008,319(5865):908-909.
[3] Chae H G,Minus M L,Rasheed A,et al.Stabilization and carbonization of gelspunpol yacrylonitrile/single wallcarbon nanotube composite fibers[J].Polymer,2007,48(13):3781 -3789.
[4] Chae H G,Choi Y H,Minus M L,et al.Carbon nanotube reinforced small diameter polyacrylonitrile based carbon fiber[J].Compos Sci Technol,2009,69(3/4):406 -413.
[5] Novoselov K S,Geim A K,Morozov S V,et al.Electric field effect in atomically thin carbon films[J].Science,2004,306(5696):666-669.
[6] Ramanathan T,Abdala A A,Stankovich S,et al.Functionalized graphene sheets for polymer nanocomposites[J].Nat Nanotechnol,2008,3(6):327 -331.
[7] Kovtyukhova N I,Ollivier P J,Martin B R,et al.Layer-bylayer assembly of ultrathin composite films from micron-sized graphite oxide sheets and polycations[J].Chem Mater,1999,11(3):771-778.
[8] Stankovich S,Dikin D A,Piner R D,et al.Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide[J].Carbon,2007,45(7):1558 -1565.
[9] Szabo T,Berkesi O,Forgo P,et al.Evolution of surface functional groups in a series of progressively oxidized graphite oxide[J].Chem Mater,2006,18(11):2740 -2749.
[10]Zhang Hailong,Xu Lianghua,Yang Fengyuan,et al.The synthesis of polyacrylonitrile/carbon nanotube microspheres by aqueous deposition polymerization under ultrasonication [J].Carbon,2010,48(3):688-695.
[11]Li Dan,Muller M B,Gilje S,et al.Processable aqueous dispersions of graphene nanosheets[J].Nat Nanotechnol,2008,3(2):101-105.
[12]Meyer J C,Geim A K,Katsnelson M I,et al.The structure of suspended graphene sheets[J].Nature,2007,446(7131):60-63.
[13]Stankovich S,Dikin D A,Dommett G H B,et al.Graphenebased composite materials[J].Nature,2006,442(7100):282-286.
