高速逆流色谱分离纯化紫甘蓝花色苷
2012-03-20易建华潘毛头朱振宝
易建华 潘毛头 朱振宝
(陕西科技大学生命科学与工程学院,陕西 西安 710021)
紫甘蓝(red cabbage)又称赤甘蓝、红甘蓝,属十字花科,是结球甘蓝的一个类型,它营养丰富,含丰富的VC,VE。研究表明:紫甘蓝花色苷具有一定的还原能力和较强的抗脂质过氧化能力,能降低血清中的脂肪含量[1],并对DPPH 自由基的清除有较强的效果[2,3]。目前,紫甘蓝花色苷的提取及纯化方法有水提取法、有机溶剂提取法、超声波提取法、大孔树脂法和凝胶过滤法等[4,5],但是这些大多数是传统方法,提取花色苷的量较少,纯度较低,也无法用高效液相色谱法(HPLC)完全分离出来一种或多种组分。
高速逆流色谱(high-speed counter-current chromatography,HSCCC)是利用物质在两相中分配系数的不同而实现分离的一种色谱技术。与传统的液-固色谱相比,它具有高效、快速、操作简单、回收率高、制备量大等的优点,已经被广泛应用于天然产物的分离纯化和食品原料的开发利用等领域[6-10]。
本试验以紫甘蓝为原料,采用超声波溶剂辅助法提取分离花色苷,并用高速逆流色谱法对花色苷进行进一步的分离纯化,得到纯度比较高的花色苷,为紫甘蓝花色苷的进一步开发利用提供依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
紫甘蓝:产地为陕西西安;
LSA-21大孔树脂:西安蓝晓科技有限公司;
甲醇、乙醇:分析纯,市售;
乙腈、正丁醇、甲基叔丁基醚:色谱纯,天津市科密欧化学试剂有限公司;
三氟乙酸:化学纯,上海科丰化学试剂有限公司;
试验用水:去离子水。
1.1.2 主要仪器设备
高效液相色谱仪:Waters2487,美国Waters公司;
紫外-可见分光光度计:UV-2600型,上海尤尼柯仪器有限公司;
高速逆流色谱仪:TBE-300A,上海同田生物技术有限公司;
树脂柱:1m×4cm,上海锦华仪器有限公司。
1.2 方法
1.2.1 超声波溶剂辅助法提取紫甘蓝中的花色苷 采用65%乙醇超声波辅助提取花色苷。取1kg 新鲜紫甘蓝,料液比1∶5(m/V),调节pH 为2.5,超声波功率为200 W,温度40 ℃,提取时间为8min,过滤,合并滤液,60 ℃下减压浓缩,得花色苷粗提液。花色苷粗提液通过LSA-21大孔树脂柱吸附处理,然后分别用3BV 的蒸馏水和3BV 30%,50%,75%的乙醇依次洗脱,流速为2BV/h,收集洗脱液,浓缩,冷冻干燥,得到紫甘蓝花色苷粉末1.73g,置于4 ℃冰箱保存备用。
1.2.2 HSCCC溶剂系统及样品溶液的制备 以正丁醇-甲基叔丁基醚-乙腈-水-三氟乙酸体积比为2∶2∶1∶5∶0.01的比例配制两相溶剂系统,摇匀使其充分混合,待两相溶液平衡后,分别置于棕色广口瓶中,其中上相为固定相,下相为流动相。
将配好的上相和下相超声脱气20min,然后称取紫甘蓝花色苷粉末300mg,溶于10mL 的上相与10mL 下相溶液中,用0.45μm 微孔滤膜过滤,备用。
1.2.3 HSCCC 的分离方法 进样前,开启循环水浴仪,将温度设定为25 ℃,以9mL/min的速度将固定相泵入主机,待固定相充满螺线管后,开启主机电源,缓慢正向调节转速至800r/min,等转速稳定后,以2mL/min的速度泵入流动相,待整个系统建立动态平衡后,将20mL 样品溶液通过进样孔注入六通阀内,开始分离,并在254nm 波长下采集数据,按照色谱峰收集流分,流分由自动接收器收集,流速设定为3mL/min,每6mL收集1管。
1.2.4 HSCCC两相分配系数的测定 根据文献[9~11],修改如下:采用HPLC测定样品在两相中的分配系数。取花色苷粗提样1~2mg,溶于2mL 上相与2mL 下相溶液中,摇匀使样品充分溶解。待分配平衡后,分离上下相,并分别量取上相和下相各1mL,用HPLC检测分析,按式(1)计算不同溶剂分配系数K 值:

式中:
K—— 化合物在两相中的分配系数;
A1—— 上相中化合物的峰面积;
A2—— 下相中化合物的峰面积。
1.3 HPLC检测分析
采用HPLC分析紫甘蓝花色苷粗提物,以及经HSCCC分离出来的各组分。色谱条件:反相C18柱(Diamonsil C18,5μm,250×4.6mm),流动相:洗脱剂(A)2%的甲酸乙腈溶液,洗 脱 剂(B)2% 的 甲 酸 水 溶 液(pH 2.57)。流 速:1.0mL/min;柱 温:20 ℃;进 样 体 积:20 μL;进 样 浓 度:1mg/mL;检测波长:525nm。
2 结果与讨论
2.1 紫甘蓝花色苷提取物制备方法的选择
目前,提取花色苷的方法一般是水提取法和溶剂提取法。但是水提法提取时间长、提取率低、杂质较多,而有机溶剂提取法消耗大、费用高、且易造成环境污染,也不适用于大量制备[4-6]。本试验采用超声波辅助溶剂提取法提取紫甘蓝花色苷,分别以乙醇浓度、提取时间、提取温度、超声波频率、料液比等为影响因素,在单因素的基础上,选择提取时间、提取温度、乙醇体积分数3种因素,各设计3个水平,通过正交试验来优化超声波辅助溶剂提取的最佳工艺条件。其正交因素水平及结果分析见表1、表2。
由表2可知,超声波辅助提取花色苷的最佳工艺条件为时间8min、温度40℃、乙醇体积分数65%,此时经超声以后的溶液其吸光度达到最大值。比较分析影响花色苷提取的3个因素,它们的影响大小顺序依次为乙醇体积分数>温度>时间。

表1 正交因素水平表Table 1 Factors and levels
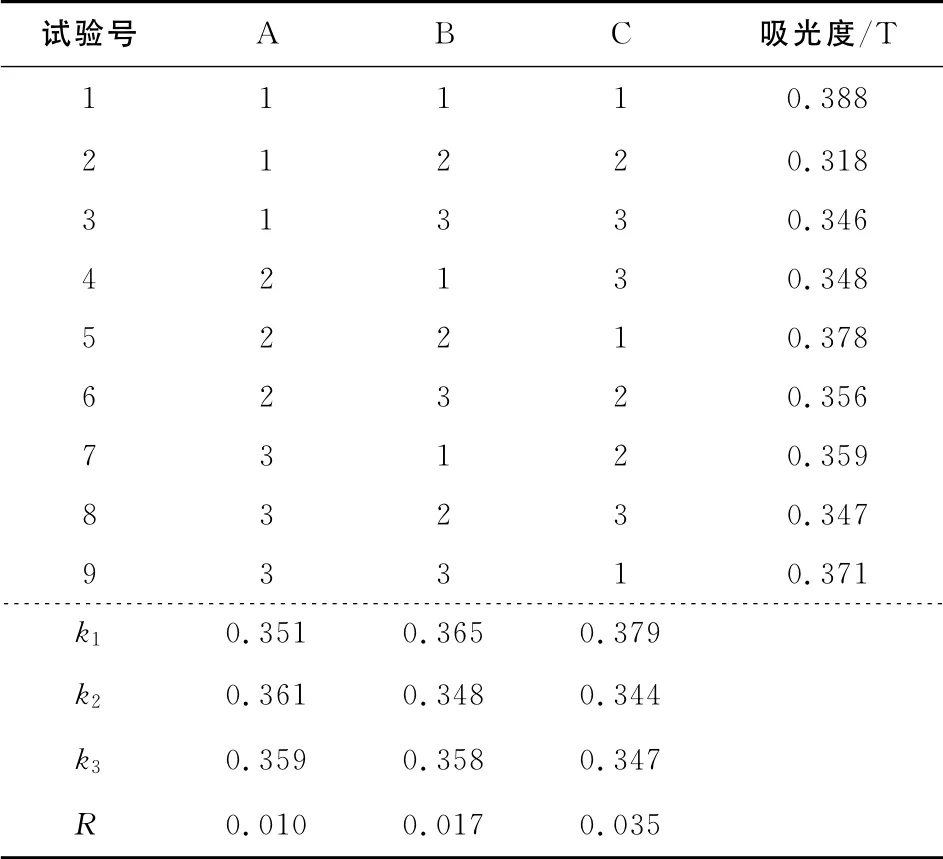
表2 正交试验结果分析Table 2 Results of the orthogonal test
2.2 HSCCC溶剂系统的研究
在HSCCC 分离的过程中,选择合适的溶剂系统是至关重要的。一般来说:合适的分配系数K 值应在0.5~2.0,并且各组分的分配系数值要有足够大的差异[12]。目前分配系数的测定多采用薄层色谱法(TLC)、毛细管电泳法(CE)、高效液相色 谱 法(HPLC)及分析型HSCCC 法[13-16]。依 据 文献[17~20],本试验采用正丁醇-甲基叔丁基醚-乙腈-水-三氟乙酸为溶剂系统,并用HPLC 法测定测定其K 值。试验依据目标组分的K 值,对正丁醇-甲基叔丁基醚-乙腈-水-三氟乙酸这一溶剂体系的不同配比进行了比较筛选,其各溶剂配比及K 值见表3。
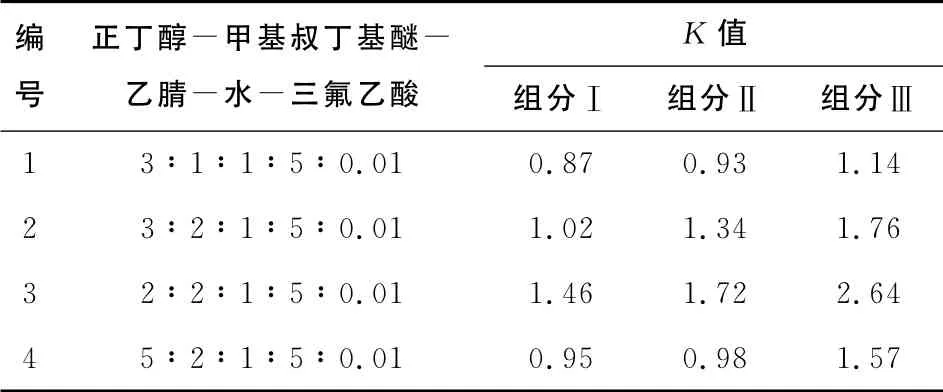
表3 不同溶剂体系配比的分配系数Table 3 Distribution coefficient(K)of compounds in solvent system with different proportions
试验考察多种溶剂体系的HSCCC分离效果。由表3可知,在以上4个溶剂系统中,各组分的分配系数K 值大都在0.5~2.0,基本符合分配系数要求,但是在溶剂体系1、2、4中,由于K 值比较接近,导致3组分不能完全分离,而溶剂体系3中的K 值相差比较大,对三目标组分有较好的分离,综合考虑实际分离效果和效率,最终选用溶剂体系3作为分离紫甘蓝花色苷的HSCCC溶剂体系。
2.3 HSCCC分离纯化的结果
按照1.2.3的方法,以正丁醇-甲基叔丁基醚-乙腈-水-三氟乙酸(2∶2∶1∶5∶0.01,V/V/V/V)为溶剂体系,分离出3 个组分,测得固定相保留率为56.6%。并且从300mg紫甘蓝花色苷粗提样中分离得到化合物Ⅰ(84mg),化合物Ⅱ(56mg),化合物Ⅲ(13mg)。HSCCC 分离图谱见图1。
由图1可知,图中有3个比较明显的峰,分别在77,105,205min时出现,这表明紫甘蓝花色苷提取样经HSCCC 分离得到3种化合物,分别命名为组分Ⅰ,组分Ⅱ,组分Ⅲ。
2.4 紫外可见全扫描
花色苷在紫外区和可见光区的最大吸收波长分别为275nm和523nm 左右[21],可以通过紫外可见扫描物质的最大波长来判定该物质是否为花色苷类物质。
将紫甘蓝花色苷粗提样和HSCCC 分离出来的3 个组分,分别用0.025mol/L 的氯化钾溶液稀释至一定浓度,用紫外可见分光光度仪进行紫外可见全扫描,结果见图2。

图1 紫甘蓝花色苷的HSCCC分离图谱Figure 1 HSCCC chromatogram of anthocyanins from purple cabbage
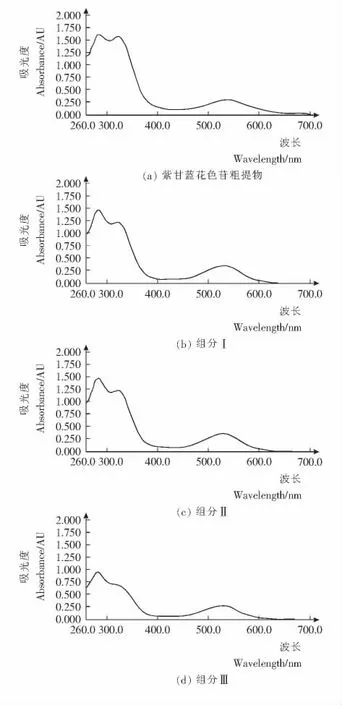
图2 紫甘蓝花色苷粗提物及HSCCC分离组分紫外可见全扫描图谱Figure 2 Ultraviolet visible full scan of anthocyanins and three components from purple cabbage
由图2可知,紫甘蓝花色苷粗体样和组分Ⅰ、Ⅱ、Ⅲ在275nm 和523nm 附近都有最大吸收峰,所以可以确定3组分均为花色苷。
2.5 HPLC检测的结果
用HPLC对紫甘蓝花色苷粗提物和HSCCC 分离所得的3个组分进行纯度检测分析,结果见图3和图4。根据峰面积计算出组分Ⅰ、Ⅱ、Ⅲ的纯度分别为76.28%,5.46%,91.46%,经过浓缩、冻干后其质量分别为84,56,13mg。
由图3可知,花色苷粗提取经过HPLC检测得到3个比较高的峰,结合3组分的HPLC图谱,可以确认,它们依次为组分Ⅲ、组分Ⅱ、组分Ⅰ。
由图4可知,3个组分经过HPLC检测,都等达到了良好的分离效果,特别是组分Ⅲ,几乎没有杂质,纯度达到了91.46%,组分Ⅰ、组分Ⅱ的纯度分别为76.28%,45.46%。
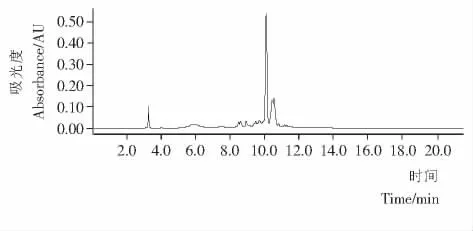
图3 紫甘蓝花色苷粗提物的HPLC图谱Figure 3 HPLC chromatogram of anthocyanins from red cabbage
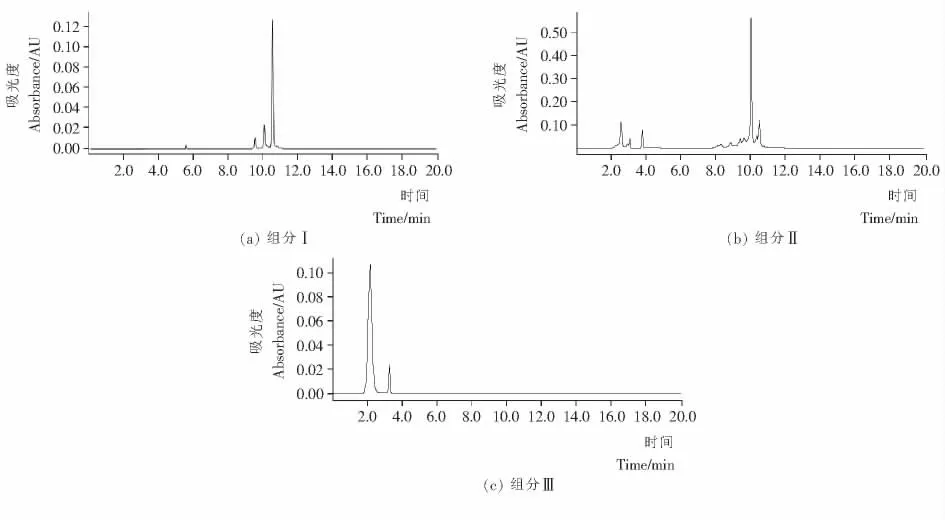
图4 紫甘蓝花色苷3个组分的HPLC色谱图Figure 4 HPLC chromatograms of three components
3 结论
采用超声波溶剂辅助提取法提取紫甘蓝花色苷,优化出最佳工艺条件为提取时间8min、提取温度40 ℃、乙醇体积分数65%。并利用HSCCC 色谱法从紫甘蓝花色苷中分离得到得到3 种化合物,300 mg 紫甘蓝花色苷提取物经过HSCCC分离后得到84 mg 纯度为76.28%的化合物Ⅰ、56mg 纯度为45.46%的化合物Ⅱ、13mg纯度为91.46%的化合物Ⅲ,但要具体确定各个分离组分的结构,还需经过核磁共振等试验来鉴定。
1 Hollman P C H,Hertog M G L,Katan M B.Analysis and health effects of flavonoids[J].Food Chem.,1996(57):43~46.
2 Sichel G,Corsaro C,Scalla M,et al.In vitro scavenger activity of some flavonoids and melanin against O2-[J].Free Radical Biol.Med.,1991(11):1~8.
3 Rice-Evans C,Miller N J,Paganga G.Structure-antioxidant activity relationships of flavonoids and phenolic acids[J].Free Radical Biol.Med.,1996,20(7):933~956.
4 于东,陈桂星.花色苷提取、分离纯化及鉴定的研究进展[J].食品发酵与工业,2009,35(3):127~133.
5 李韬,张宏宇.花色苷类色素的研究进展[J].农业科技与装备,2010(5):23~27.
6 伍方勇,戴德舜,王义明.高速逆流色谱与质谱联用在中药分析中的应用[J].高等学校化学学报,2002,23(9):1 698~1 700.
7 Sutherland I A.Recent progress on the industrial scale-up of counter-current chromatography[J].Journal of Chromatography A,2007,1 151(1~2):6~13.
8 高冷,谢冠.高速逆流色谱分离纯化冻青中的黄酮类化合物[J].化学与生物工程,2012,29(2):73~75.
9 程悦,严志勇.高速逆流色谱分离制备苦茶中的苦茶碱[J].中山大学学报(自然科学版),2010,49(3):65~69.
10 郭丽丽,刘景圣.高速逆流色谱实验在有效成分分离中溶剂体系的选择[J].农产品加工,2010(5):27~29.
11 李艳,肖凯军.高效逆流色谱研究进展-在天然产物有效成分分离方面的应用[J].现代食品与药品杂志,2006(4):78~80.12 李佳银,罗晋.高速逆流色谱法分离紫锥菊花色苷及其抗氧化性研究[J].分析测试学报,2012,31(1):45~50.
13 时新刚,陈志伟.高速逆流色谱应用研究进展[J].生命科学仪器,2009(8):3~5.
14 李玉兰,张小燕.黄芩中总黄酮的提取及高速逆流色谱分离纯化[J].中药新药与临床药理,2010,21(4):437~439.
15 Kamaljit Vilkhu,Raymond Mawson.Applications and opportunities for ultrasound assisted extraction in the food industry-A review[J].Innovative Food Science and Emerging Technologies,2008(9):161~169.
16 Ito Y A,Conway W D.High-speed counter-current chromatography[M].New York:Wiely/Interscience,1996:36.
17 A Degenhardt,H Knapp,P Winterhalter.Separation and purification of anthocyanins by high-speed countercurrent chromatography and screening for antioxidant activity[J].Agric Food Chem.,2000,48(2):338~343.
18 Michael Schwarz,SiIke Hillebrand,Saskia Habben,et a1.Application of high-speed countercurrent chromatography to the large-scale isolation of anthocyanins[J].Biochemical Engineering Journal,2003(14):179~189.
19 Torskangerpol K,Chou E,Andersen O M.Separation of acylated anthocyanin pigments by high speed CCC[J].Liq.Chrom.&Rel.Technol.,2001,24(11&12):1 791~1 799.
20 Du Qizhen,Gerold Jerz,Peter Winterhalter.Isolation of two anthocyanins sambubiosides from bilberry(vaccinium myrtillus)by high-speed countercurrent chromatography[J].Journal of Chromatography A,2004(1 045):59~63.
21 孙建霞,张燕,孙志健,等.花色苷的资源分布以及定性定量分析方法研究进展[J].食品科学,2009,30(5):263~268.
