浅析无菌药品生产中空气阻断的内涵
2012-02-27邓海根
邓海根 张 华
(1. 无锡华瑞制药有限公司,江苏无锡 214062;2. 上海市食品药品监督管理局认证审评中心,上海 200020)
1 引言
欧盟无菌药品附录2008第50条对无菌生产提出了一些特殊要求,其中十分重要的一条是采用空气阻断,防止倒灌。WHO 2010无菌药品中也有类似提法。在欧盟及WHO无菌药品原文出现了几个特有的名词,如Air break;Drains;Floor drains;Trap或water seals,我国制药行业内对此虽有译文,然而它们的技术内涵较深,值得人们用质量风险管理的理念去深思,以避免设计和改造中的失误。
2 历史及法规的简要回顾

图1 70年代注射剂灭菌柜排水管路连接示意图
在七十年代,美国曾爆发了一系列的败血症病例。1971年3月第一周内,美国7个州8个医院发生了150起败血症病例。一周后,败血症病例激增至350人,截至1971年3月27日,总数达到405个病例。污染菌为阴沟肠杆菌(Enterobactercloacae)或欧文氏菌(Erwinaspp)。1972年,英国德旺波特(Devonport)医院使用被污染的葡萄糖输液导致6起败血症死亡病例[1]。FDA成立了特别工作组,对此作了长达一年多的调查发现,危及安全的高风险因素是多方面的,如:配液罐、灭菌柜、过滤器清洗池等与地漏直接相接、灭菌不完全等。例如,灭菌柜的排水管路直接与阴沟相连(图1),阴沟肠杆菌、欧文氏菌等在灭菌的冷却阶段随着污水倒灌入灭菌柜,污染产品密封口,使用过程中穿刺将污染菌带入产品,可导致败血症。至于过滤器及配液罐的潜在风险,将在下节讨论。
为了消除无菌药品生产中这类高风险因素,FDA开始强调无菌药品灭菌过程对任何与产品或物品相接触的冷却介质的微生物控制,与此同时,Air break(空气阻断)也进入美国FDA的行业指南并频频出现在其它技术文献中。
在欧盟无菌药品附录2008中,第50条规定:无菌生产的A/B级区内禁止设置水池和地漏。在其它洁净区内,机器设备或水池/水槽与地漏不应直接相连。在洁净要求较低的区域,地面排水设施应设隔离装置或水封,防止倒灌。值得注意的是空气阻断的应用范围扩大到了设备(如:配液罐、灭菌柜等)以及水槽(如过滤器超声波清洗池等)。WHO无菌药品指南2010提法雷同。空气阻断是防止无菌药品污染的极为重要措施,意思是指上述生产设备不要与下水道直接相连。
ASME BPE在国际标准中,对空气阻断以下图表述[2],这种形象化的处理方式对理解空气阻断的设置是十分有益的。
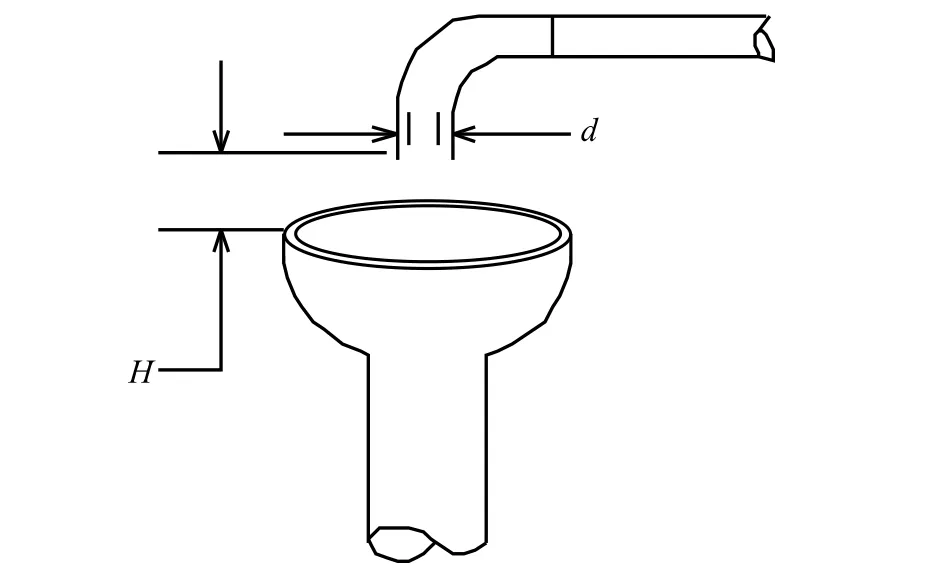
图2 空气阻断示意图

3 所见安装缺陷及其风险
在无菌操作区,不会出现水池及地漏方面的缺陷,因为这个区域不允许设计水池及地漏。然而,无菌药品接受罐、配制罐等的连接方式往往出现潜在的风险。这与对空气阻断正确理解与否直接相关。其中,无菌药品的配制区出现的问题较为突出,现以此为例加以讨论。这类缺陷大体上可用示意图3来表示。
图3中,A、B、D、E、H为阀门,其中E可能是止回阀,企业的意图是防止倒灌,E-F之间相当于水封,防止外部气体进入洁净区。F-G之间则直接相连。
可以理解,药液配制完成后,D关闭,药液经A和B阀送往下道工序—过滤或灌装;在实际生产线上,这种药液贮罐还可能是无菌药液的接收罐,安装在无菌操作区,从主观愿望上说,E-F的设置是避免B+A区出现地漏。

图3 缺陷安装连接示意图
当然,上述安装方式也可能是C级区的配液罐。当贮罐在清洗时,清洗液/注射用水走A、D、E、F,然后通往总排水管G排放。
当对贮罐进行在线灭菌时,D阀关闭,疏水器C用于节约蒸汽,排放冷凝水和气体。
这一安装方式的风险在于E-F之间积水长菌,在带抽真空、工艺过程有热胀冷缩现象的设备上,如无空气阻断的安装方式,就有可能出现倒灌,出现上一节提到的严重风险。
其次,如果E是止回阀,这种设置不符合优良设计规范的要求。众所周知,在线灭菌需要疏水器的有效运行,将气体和冷凝水尽快排放干净,而止回阀的安装对实现这一目标有不利影响。
现以图4来进一步分析质量的风险。如图所示,不同房间的排水,一般采用同一总排水管路。而不同的贮罐可能安装在不同房间,或在同一室内。安装高度可能不同,如楼上楼下,水压会有差异。当T1已清洁待用时,底阀可能不关,以排尽积水,此时,如总排水管管径偏小,或因故堵塞,或设备的液位差过大等原因,T2、T3清洁时大量的清洗水可能将总排水管路中的污水一起压入已清洁的贮罐T1,在人们没有察觉的情况下造成了微生物污染和产品交叉污染;过滤器清洗池、贮罐抽真空、温差变化等也可能导致类似的风险。可见,从避免风险角度来看,国际及国内规范条款的要求是十分必要的。
现将图4中的T1换成清洗池(图5),如药液过滤器清洗池(左)或灌装机部件清洁池(右),出现倒灌后果所致的风险也是不言而喻的。
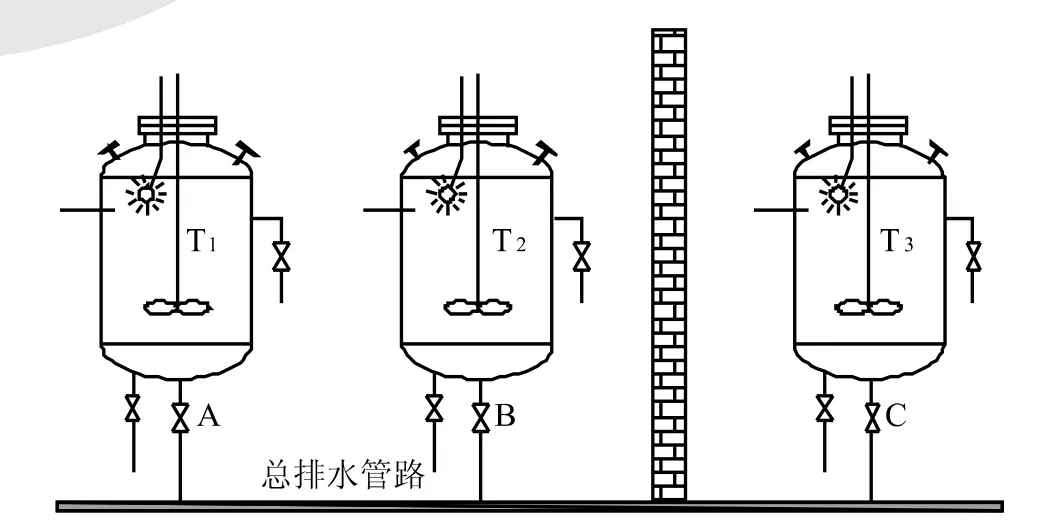
图4 污染及交叉污染风险连接示意图

图5 直接接触产品的器具清洗池示例
4 对国际规范的理解及相关名词的解释
如将对欧盟无菌药品第50条的理解,画成示意图(图6)来表述设备与地漏之间的关系,对读者也许会有所帮助。
什么是Air break?Air break=空气阻断=不直接相连,阻断的位置是在设备、水槽等与地漏之间,欧盟用了设备(machine)或水槽(sink)这样的词,十分明确,通常不会引起误解。

图6 对国际规范理解示意图
更衣室中洗手池并不会引起质量的风险,因此,设地漏带水封即可,不必采用空气阻断方式。生产线相关的废液罐,例如存经灭活或中和后即可排放废液的贮罐,也可根据实际风险情况,不一定采取空气阻断的形式,可采用专用排水阀的形式。
与此相关,以前学术界在讨论中,在很多场合下谈到Floor drains的译法,将它译成明沟,笔者认为这种译法并不十分确切,因为在药品生产的C、D级,是不太可能设置明沟的。图7是对地面排水设施的图示注释。可以理解,如果需清洗的设备较大,那就可能有必要采取Floor drains(地面排水设施)这种形式。例如:将可移动式配制罐、桶放在栅板上,进行清洁,水经过二层栅板排出,排水设有水封[注1]注1 :照片中黑色部分由边框不平折光所致,并非霉斑。。这样的形式显然并不是人们通常所理解的明沟。然而,正如美国机械工程师协会的“生物工艺设备”一书中所示,对注射用水及纯化水系统而言,它们的贮罐不设在洁净生产区,而系统出水或回水的取样点或系统排空阀一般设在制水间,此时sink可以是明沟,而这类阀门均不得与地漏直接相连(参见图8),即采用物理隔断的做法(physical break以及air gap)。
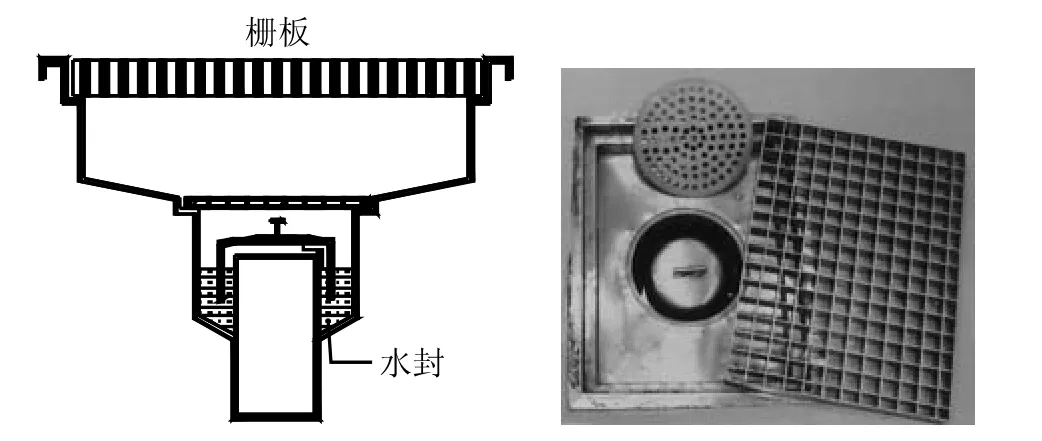
图7 地面排水设施示意图
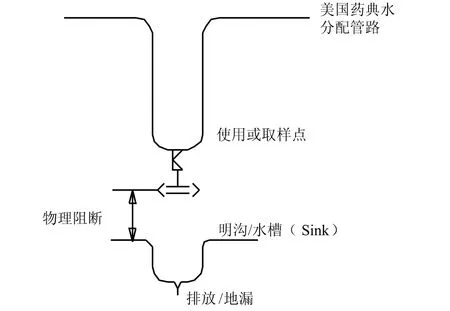
图8 水系统的防污染措施
顺便提一下,在欧盟50条中,还有二个词“Trap或water seals”,这是指可以让水通过,但又能防止下水道气体倒流的U形或S形隔臭管,即是人们常说的水封。
5 企业的担忧和建议的解决方案
出现第三节中提到的缺陷,主要原因是对国际化规范的背景了解不够,美国生物工艺设备[注2]注2 :ASME Biopharmaceutical Processing Equipment不一定指生物制品的生产设备,可理解为卫生工艺设备。对在线灭菌管路连接要求(图9),对理解国际标准也许是有帮助的;另一个客观原因是企业担心设备在线灭菌时,有部分水汽留在洁净区,造成湿度偏大,易于长菌,尽管无菌药品生产中,在线灭菌未必要每天/班进行。

图9 设备纯蒸汽灭菌管路的空气阻断示意图
大容量注射剂用的大型蒸汽灭菌柜,通常设在一般区,湿度问题不必过于担心。贮罐和脉动真空灭菌柜,有可能设在C或D级区,B级区的无菌药液贮罐的排放管需接向C或D级区,或采用其它手段,避免对B级区构成污染的风险,C或D级区设备在线灭菌完成时蒸气的尾气可在设备的上方排放,例如:设止回阀或其它方法,需视企业的具体情况而定。
生产设备使用中往往需要在线清洁及在线灭菌,需要排水及排水汽,图10A及B进一步给出了简化的示意方案。
A及B二图中的PS(pure steam)意为纯蒸气,纯蒸气的冷凝水由在线灭菌形成。PS冷凝水以上的设备就是欧盟GMP中提到的machine(配液罐等生产设备,为了将图简化,设备、通产品的管路和相应的阀门均没有标出)。但图表出了设备关键的连接方式:有空气阻断且有水封。从图可看出,设备在线灭菌时,冷凝水和不凝性气体经疏水器排放;在线清洁时,则可通过右侧的旁路排放。图A所示设备较小,少量的冷凝水及蒸气可直接排放入生产区;而B的设备比较大,采用了换热器的方式,来降低夹带蒸汽的冷凝水在排放时对生产区环境的影响。换言之,换热器及温度控制只在必要时才设。有些企业将排水温度控制在45℃左右,一则对人员安全,二则当蒸气变成冷凝水时,体积缩小了1 700倍,有利生产环境的保持。而图10B中冷却水还可以是生活热水的进水(节能、综合利用),总之,企业的担忧有各种可选的解决方案。
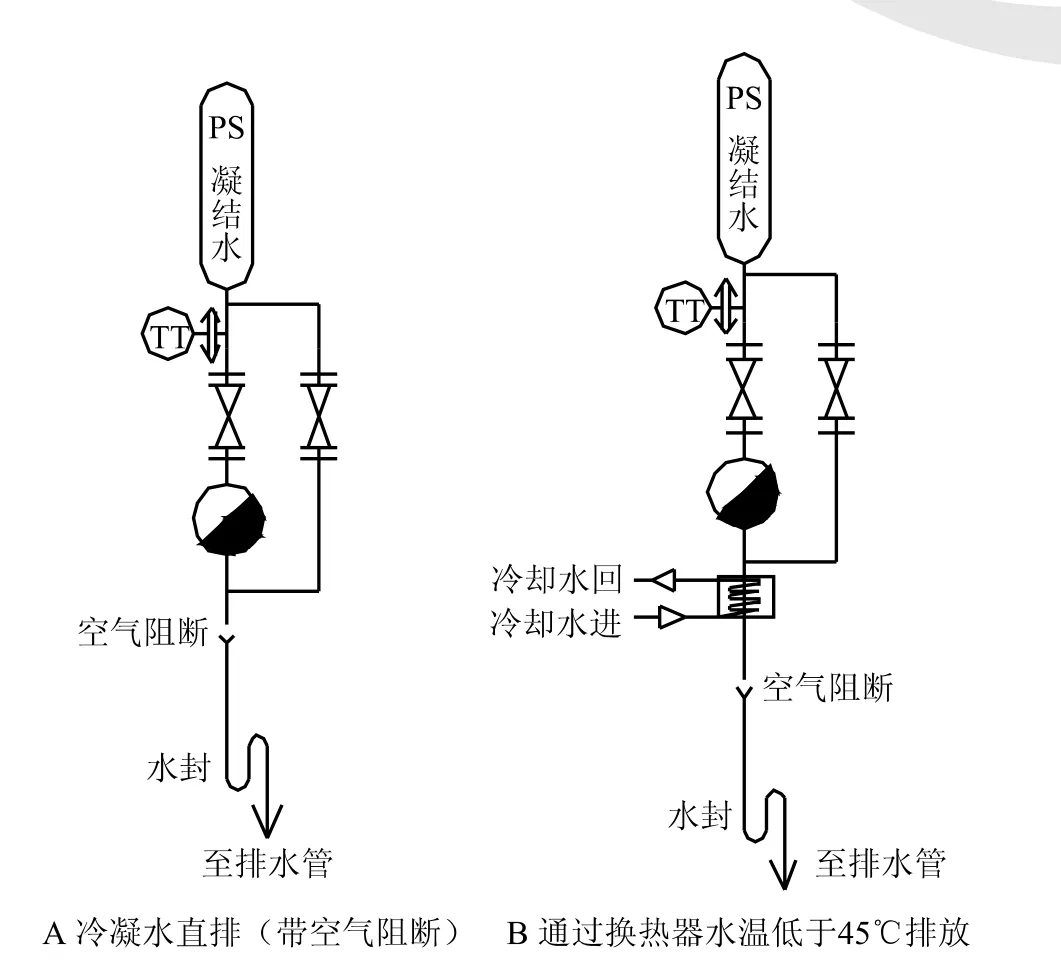
图10 生产设备排放防污染方式示意图
6 小结
药品GMP的法规条款充分考虑了影响产品质量的风险,空气阻断是无菌药品生产中国际上采用的通用标准。
无菌药品生产企业将需在线清洁及在线灭菌的设备直接接入地漏,主要是担心SIP过程中蒸气对生产环境的影响,然而,这种对环境的风险与生产设备接入地漏相比,明显处于从属地位,而且有多种解决方案可供选择。
要执行好法规条款,有必要对法规条款的目的及背景作深入的了解,并对本企业的风险进行评估。
[1] FDA's Proposed GMPs for large Volume Parenterals (LPV) “ The Gold Sheet” Vol. 10.No 5. May 1976.
[2] ASME Bioprocessing Equipment 2009.
[3] Bernard T. Loftus Robert A. Nash Pharmaceutical Process Validation. 1993.
[4] 中国化学制药工业协会,中国医药工业公司主编.药品生产验证指南[S]. 北京:化学工业出版社,2003.
[5] 欧盟GMP无菌药品附录-2008.
[6] WHO GMP 无菌药品-2010.
[7] 我国GMP规范 无菌药品附录-2011.
[8] PDA Technical Report No. 40 Sterilizing Filtration of Gases 2005
