镍基双金属催化剂上甲烷脱氢反应的密度泛函理论
2012-01-10朱贻安周兴贵
范 琛,朱贻安,周兴贵
(华东理工大学化学工程联合国家重点实验室,上海 200237)
镍基双金属催化剂上甲烷脱氢反应的密度泛函理论
范 琛,朱贻安,周兴贵
(华东理工大学化学工程联合国家重点实验室,上海 200237)
为了从原子尺度上研究双金属的作用机理,采用基于密度泛函理论的第一性原理方法研究了甲烷在 Ni-Pt和Ni-Pd双金属催化剂上的脱氢过程。计算结果表明,在Ni催化剂中引入Pt或Pd会大大减弱表面C原子的吸附,从而提高催化剂的抗积炭能力,而且甲烷第一步脱氢活化能会有所提高,反应活性略有降低。
密度泛函数理论 镍 双金属催化剂 甲烷 脱氢
甲烷的脱氢反应是很多重要化工反应的关键步骤,如甲烷的直接裂解制备纳米碳纤维[1]、甲烷的蒸汽重整反应[2]和甲烷的干气重整反应[3]等。因此,甲烷的脱氢反应无论是在催化领域还是化学工业中都是一个很值得研究的反应体系。负载型的Ni催化剂是这一反应过程中最常见的催化剂,但Ni催化剂表面非常容易形成积炭,表面碳原子会进一步形成纤维状的石墨结构,包裹催化剂表面使其失活。因此,提高催化剂的抗积炭能力,延长催化剂的使用寿命是该催化剂制备的研究重点,也是甲烷重整反应中一直存在的问题和面临的难点。一般,通过在Ni催化剂中引入金属助剂形成合金催化剂,来改变催化剂的催化性能,从而提高催化剂稳定性,如,在Ni催化剂中添加Pt和Pd可以显著提高催化剂的抗积炭性能[4,5]。然而,目前对这些金属助剂影响Ni催化剂性能的机理尚不清楚。为了研究金属助剂和Ni催化剂之间的作用机理,本工作采用基于密度泛函理论的第一性原理计算方法,研究了Ni-Pt和Ni-Pd双金属催化剂上的甲烷脱氢反应,为筛选高效的甲烷脱氢催化剂提供理论依据。
1 计算方法和模型
本工作采用VASP软件进行计算[6,7]。该软件包中,周期性体系内的每个k点的波函数可以看作是一个波形部分和一个周期性部分的乘积,其中后者可以用一个平面波基组展开。平面波基组的能量阈值是400 eV。价电子和离子实之间的相互作用通过Blöchl的全电子投影缀加波(PAW)方法进行描述[8]。交换相关泛函采用基于广义梯度近似(GGA)的PBE函数[9]。采用Monkhorst-Pack网格在第一布里渊区内取样[10],由Methfessel-Paxton方法决定电子占据,smearing的宽度为0.2 eV[11]。由于在体系内包含了具有磁性的Ni元素,因此在计算中考虑了自旋极化效应。前人的工作[3]表明,金属表面的磁性对于准确地计算体系的总能十分重要。
通过计算Pt和Pd两种金属在Ni(111)表面的偏析能(Eseg)来确定Pt,Pd与Ni形成合金后的微观结构。偏析能的计算公式为:

其中:Es为第二组分金属原子取代表面一个Ni原子的总能;Eb为第二组分金属原子取代体相中一个Ni原子的总能。由此得到的Eseg若为负值,则表示第二组分金属倾向于在Ni表面形成表面合金;反之,若为正值,则表示容易形成体相合金。对于Pt和Pd来说,计算出的偏析能均为负值,说明这两种金属比较容易和Ni形成表面合金,因此,在本研究的计算模型采用表面合金的模型。
Ni(111)的计算模型由包含四层Ni原子的p(2×2)的周期性平板构成。为了检验模型大小对吸附的影响,分别计算了p(2×2),p(3×3)和p(4×4)晶胞中碳原子的吸附能,分别为-6.87,-6.86和-6.88 eV。这表明p(2×2)已足够消除吸附质的侧面相互作用。构建M/Ni(111)(M代表Pt或Pd)表面时,将Ni(111)表面上的一个Ni原子替换为一个M原子即可。采用5×5×1的k点网格在第一布里渊区取样。在计算过程中,底部两层原子固定,顶部两层原子和吸附能允许弛豫。平板模型之间以1.2 nm的真空层隔开以消除周期性相互作用。
甲烷脱氢反应的中间体CHx(x为0~3)的吸附能的计算公式为:

甲烷脱氢反应中所有的过渡态(TS)是用dimer方法搜索的[12]。采用共轭梯度法来优化所有的构型[13],当所有方向上的力小于3×108eV/m,得到相应的极小点或鞍点。为了得到力的准确结果,在进行电子优化时系统的总能量收敛到每摩尔原子1×10-7eV。
2 结果与讨论
2.1 反应中间体的吸附
CHx在Ni表面上会优先吸附在三齿空位[3]。和Ni(111)相比,双金属催化剂表面上由于引入了新的金属原子,破坏了表面的均一性,会有几种不同的三齿空位。根据三齿空位中局域原子组成的不同,这些空位大体可以分为两种:一种是由三个Ni原子组成,记为Ni3位;另一种由两个Ni原子和一个M原子组成,记为Ni2M位。
对于CH3、CH2和CH的最稳定吸附位为Fcc位(指组成表面三齿空位的原子正下方有一个原子位于模型的第三层),而C原子的吸附位为Hcp位(指组成表面三齿空位的原子正下方有一个原子位于模型的第二层)。吸附质CHx在双金属表面上的吸附构型和Ni(111)表面上的吸附构型比较相似,其结果如图1所示。由图可知:对于C吸附模型,C原子位于Hcp位的中心位置;对于CH的吸附构型,C原子位于Fcc的中心位置,并且C-H键垂直于表面指向表面外;对于CH3和CH2吸附模型,分别形成3个和1个C-H-Ni的三中心键,这些三中心键会使吸附更加稳定。同时还计算了CH4分子在所有表面上的吸附,结果发现CH4的吸附能非常小,在所有的表面上均为-0.02 eV左右,表明CH4的吸附属于物理吸附,CH4分子与金属表面之间的相互作用为范德华作用力。
比较图1中CHx在Ni3位和Ni2M位的吸附能,可以发现,在不同的双金属表面上,CHx在这两个吸附位的吸附能变化明显。在Pt/Ni(111)上,CHx在Ni2Pt位的吸附能要高于Ni3位。这表明Pt原子本身要比Ni原子更加活泼,CHx更加倾向于与Pt形成化学键,而实际上,Pt本身也是很好的甲烷活化的催化剂,具有很高的活性。由图1还可以看出,CHx在Pd/Ni(111)表面的Ni2Pd位的吸附能要低于Ni3位的吸附能。这是由于Pd具有完全占据的d带,活性比Ni要低,CHx更加倾向于与Ni形成化学键。可见,在双金属催化剂上,不同的原子组成的吸附位上的吸附能差别较大,而这种差别取决于组成吸附位的原子自身的活性,即活性比较高的原子组成的吸附位上的吸附能也较高,反之亦然。
比较CHx在不同双金属表面Ni3位以及Ni(111)上的吸附能可发现,虽然不同表面上的Ni3位的局域原子组成完全相同(均由3个Ni原子组成),CHx的吸附能却有很大的差别,如C原子在Pt/Ni(111)上Ni3位的吸附能为-6.40 eV,在Pd/Ni(111)上Ni3位的吸附能为-6.71 eV,而在Ni(111)上(所有的吸附位均为Ni3位)的吸附能为-6.87 eV。由此可见,虽然吸附位的局域原子组成完全相同,但是由于吸附位周围原子的不同,也会导致吸附能有较大的差异。造成这种差异的主要原因是Ni原子和周围的M原子之间的相互作用,改变了Ni原子的电子结构,从而导致Ni3位的吸附能发生变化。
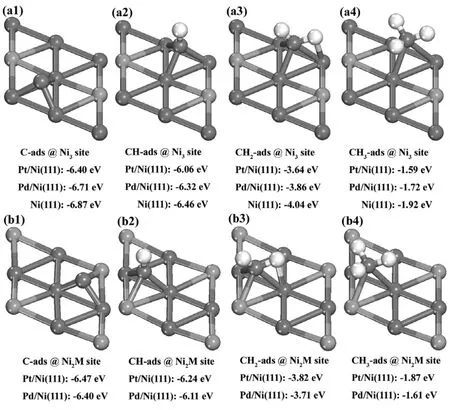
图1 CHx在Pt/Ni(111)和Pd/Ni(111)上的吸附构型和吸附能Fig.1 Adsorption configurations and adsorption energies of CHx on M/Ni(111)
2.2 双金属表面上的CH4脱氢反应
目前,学术界普遍认为,甲烷脱氢反应中第一个C-H键的断裂生成吸附态的CH3和H原子是整个过程的速率控制步骤[14,15]。因此,在研究Pt/Ni(111)和Pd/Ni(111)双金属催化剂上CHx吸附的基础上,进一步计算CH4在这两个表面上的第一步脱氢反应活化能。由于双金属表面存在两种活性位,一种完全由Ni原子组成,记为Ni位;另一种有M原子参与,记为M位。在过渡态构型中,CH3位于Atop位,而解离的H原子位于相邻的三齿空位。搜索到的过渡态构型和Ni(111)上的过渡态构型非常接近,结果如图2所示。由图可知,在Pt/Ni(111)上的活化能为1.16 eV,Pd/Ni(111)上的活化能为1.09 eV,而Ni(111)上的活化能为0.92 eV。说明两种双金属上的活化能均高于Ni(111)上的活化能。这是由于CHx在Pt/Ni(111)和Pd/Ni(111)上的Ni3位上的吸附弱于Ni(111),吸附的减弱不利于脱氢反应的进行。由图2还可以发现,Pt/Ni(111)上M活性位的活化能比Ni活性位上的活化能略低。这与上述吸附能结果一致,即解离生成的CH3在Pt/Ni(111)上Ni2Pt位的吸附强于Ni3位,而吸附的增强有利于脱氢反应的进行。相反,在Pd/Ni(111)上解离生成的CH3在Ni2Pd位的吸附要弱于Ni3位,Pd/Ni(111)表面上M活性位上的活化能高于Ni活性位上的活化能。
2.3 双金属催化剂抗积炭性能
Ni催化剂作为CH4脱氢最常用的催化剂,存在的最大问题就是容易形成表面积炭,从而导致催化剂结焦失活。为了提高Ni催化剂的抗积炭性能,通常在催化剂中加入一定量的金属助剂,形成Ni基双金属催化剂,在不损失催化剂活性的前提下,同时抑制表面积炭的形成。因此,对于设计高性能的CH4脱氢催化剂有两点至关重要,即催化剂的活性和催化剂的抗积炭能力。
CH4的第一步脱氢生成CH3是整个反应的速率控制步骤,因此,这一步反应速率的快慢决定了Ni催化剂的反应活性。一方面由于不同催化剂表面上同一个基元反应的指前因子可以认为大致相同,由阿伦尼乌斯方程可知,在相同反应温度下,反应速率仅由活化能所决定,因此,Ni基催化剂的活性可由这一步的脱氢活化能来表征。另一方面,由于C原子的吸附越强,则表面C的覆盖率也会越高,而且生成的表面C原子很难参与进一步的反应,转化成最终产物,因此,催化剂的抗积炭能力可由C原子在表面的吸附能来表征,
综上所述,C原子在Pt/Ni(111)上最稳定的吸附位为Ni2Pt位,吸附能为-6.47 eV;C原子在Pd/Ni(111)上最稳定的吸附位为Ni3位,吸附能为-6.71 eV;而C原子在Ni(111)上最稳定吸附位的吸附能为-6.87 eV(见图1)。说明与Ni(111)相比,Pd/Ni(111)和Pt/Ni(111)这两种双金属表面具有较好的抗积炭性能。同时,还发现甲烷的第一步脱氢反应的最低活化能为1.09 eV,比Ni(111)上的0.92 eV略高。考虑到CH4脱氢反应一般都在高温条件下发生,随着温度的升高,活化能对反应速率的影响会逐渐减弱。对于甲烷脱氢反应来说,反应通常在高温下进行,反应速率非常快,反应活性的略微降低并不会影响催化剂的应用,而抗积炭性能的显著提高则会大大延长催化剂的使用寿命。因此,这两种双金属催化剂都是比较理想的CH4脱氢催化剂。为了进一步验证Pt和Pd的引入对催化剂抗积炭性能的影响,计算了Ni(111), Pt/Ni(111)和Pd/Ni(111)上最后一步脱氢反应(CH→C+H)的活化能,分别为1.39,1.73和1.48 eV。这表明Pt和Pd的引入会导致最后一步脱氢反应的活化能升高,不利于表面C原子的生成,从而提高了催化剂的抗积炭性能。这一结论和An等[16]在Cu/Ni(111)上的计算结果类似。
3 结 论
a)采用基于密度泛函理论的第一性原理方法研究了Ni-Pt和Ni-Pd双金属催化剂上的甲烷脱氢反应,发现,CHx在不同Ni2M位的吸附能和吸附位的局域原子组成相关,不同的原子组成造成吸附能的差别很大,活性比较高的原子组成的吸附位上的吸附能也较高,反之亦然。另一方面,由于表面Ni原子和周围的M原子之间的相互作用改变了Ni原子的电子结构,从而导致CHx在不同Ni3位的吸附能有很大差异。
b)CH4的第一步脱氢生成CH3是整个过程的速率控制步骤,在Ni-Pt和Ni-Pd双金属催化剂上甲烷的第一步脱氢反应的活化能比Ni(111)上的略高,催化剂活性略有降低,但双金属催化剂上甲烷的最后一步脱氢反应的活化能也比Ni(111)上的高,不利于表面C原子的生成,从而提高了催化剂的抗积炭性能。而CH4脱氢反应通常在高温下进行,反应速率非常快,反应活性的略微降低并不会影响催化剂的应用,而抗积炭性能的显著提高却大大延长催化剂的使用寿命。因此,两种双金属催化剂都是较理想的CH4脱氢催化剂。
[1]Sun Y F, Sui Z J, Zhou J H, et al. Catalytic decomposition of methane over supported Ni catalysts with different particle sizes[J]. Asia-Pacific Journal of Chemical Engineering, 2009, 4(5): 814-820.
[2]Bengaard H S, Nørskov J K, Sehested J, et al. Steam reforming and graphite formation on Ni catalysts[J]. Journal of Catalysis, 2002, 209(2): 365-384.
[3]Zhu Y A, Chen D, Zhou X G, et al. DFT studies of dry reforming of methane on Ni catalyst[J]. Catalysis Today, 2009, 148(3-4): 260-267.
[4]Garcia-Dieguez M, Pieta I S, Herrera M C, et al. Nanostructured Pt-and Ni-based catalysts for CO2-reforming of methane[J]. Journal of Catalysis, 2010, 270(1): 136-145.
[5]Takenaka S, Shigeta Y, Tanabe E, et al. Methane decomposition into hydrogen and carbon nanofibers over supported Pd-Ni catalysts:Characterization of the catalysts during the reaction[J]. Journal of Physical Chemistry B, 2004, 108(23): 7656-7664.
[6]Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J].Computational Materials Science, 1996, 6(1): 15-50.
[7]Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B,1996, 54(16): 11169-11186.
[8]Blöchl P E. Projector augmented-wave method[J]. Physical Review B, 1994, 50(24): 17953-17978.
[9]Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[10]Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192.
[11]Methfessel M, Paxton A T. High-precision sampling for Brillouin-zone integration in metals[J]. Physical Review B, 1989, 40(6): 3616-3621.
[12]Henkelman G, Jonsson H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives[J].Journal of Chemical Physics, 1999, 111(15): 7010-7022.
[13]Sheppard D, Terrell R, Henkelman G. Optimization methods for finding minimum energy paths[J]. Journal of Chemical Physics, 2008,128(13): 134106.
[14]Lee M B, Yang Q Y, Tang S L, et al. Activated dissociative chemisorption of CH4on Ni(111): observation of a methyl radical and implication for the pressure gap in catalysis[J]. Journal of Chemical Physics, 1986, 85(3): 1693-1694.
[15]Lee M B, Yang Q Y, Ceyer S T. Dynamics of the activated dissociative chemisorption of CH4and implication for the pressure gap in catalysis:a molecular beam-high resolution electron energy loss study[J]. Journal of Chemical Physics, 1987, 87(5): 2724.
[16]An W, Zeng X C, Turner C H. First-principles study of methane dehydrogenation on a bimetallic Cu/Ni(111) surface[J]. Journal of Chemical Physics, 2009, 131(17): 174702-174711.
Density Functional Theory Study of Methane Dehydrogenation on Ni-based Bimetallic Surfaces
Fan Chen, Zhu Yi’an, Zhou Xinggui
(State Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China)
In order to gain insight of this process at atomic level, first-principles based on density functional theory was used to investigate methane dehydrogenation on Ni-Pt and Ni-Pd bimetallic surfaces. The calculation results showed that the introduction of Pt and Pd decreased the C adsorption energies on the bimetallic surfaces,which improved the coke resistance of the catalysts. Meanwhile, the activation energies for the first dehydrogenation step increased, indicating the decrease of the activity of the bimetallic catalysts.
density functional theory; nickel; bimetallic catalyst; methane; dehydrogenation
TQ032;TQ426.82 文献标识码:A
1001—7631 ( 2012 ) 02—0117—05
2012-02-17;
2012-03-26
范 琛(1985-),男,博士研究生;周兴贵(1966-),男,教授,通讯联系人。E-mail: xgzhou@ecust.edu.cn
中央高校基本科研业务费(WA1014027)
