鱼腥藻PCC 7120染色体上relBE同源基因的克隆及表达
2012-01-03陈思礼
陈思礼, 梅 菊
(1 中南民族大学 生命科学学院,武汉 430074;2 华中科技大学 环境科学与工程学院,武汉430074)
细菌的毒素-抗毒素系统(TA)由位于同一操纵子中的有数碱基重叠的2个基因组成,前者编码毒素蛋白,后者编码抗毒素蛋白[1],毒素蛋白可通过影响DNA 复制、mRNA稳定性、蛋白合成、细胞壁生成以及 ATP形成过程抑制细胞生长甚至导致细胞死亡[2,3].E.coil染色体上的relBE操纵子中relB和relE基因分别编码不稳定的抗毒素蛋白relB和稳定毒素蛋白RelE,RelE毒性的激活依赖于蛋白酶Lon对RelB的降解,relB的同源基因存在于许多革兰氏阳性和阴性的细菌染色体上[4,5].
水华鱼腥藻(Anabaena, 属Nostoc )是造成淡水水体水华污染的主要藻种之一,藻类水华污染的水体可导致鸟类等动物和水生生物以及人类疾病[6].当前对于细菌染色体上的毒素-抗毒素系统的生理功能存在不同的观点,有关蓝细菌中TA系统的研究报道不多见.本文通过分子生物学方法成功克隆并表达了鱼腥藻PCC7120染色体基因asl4561和asl4562,为后续纯化并研究两蛋白之间的相互作用及其在鱼腥藻PCC7120体内的生理作用奠定基础.
1 材料与方法
1.1 材料与试剂
鱼腥藻sp.PCC7120购自中国科学院水生物研究所,E.coilDH5α和E.coilBL21(DE3)为本实验室保存,pMD18-T Vector为Takara公司产品,表达载体pET28a(+)为本实验室保存. DNA聚合酶为Fermentas公司产品,限制性内切酶、T4 DNA连接酶等为Takara公司产品,琼脂糖凝胶回收试剂盒及质粒DNA提取试剂盒均为Axygene产品. PCR引物由南京金斯瑞生物科技有限公司合成,克隆片段的测序由南京金斯瑞生物科技有限公司测序确定.
1.2 实验方法
1.2.1 PCC7120基因组DNA提取
参照文献[7]方法提取基因组DNA.
1.2.2asl4561和asl4562基因的克隆
分子生物学操作按照标准方法进行[8].以PCC7120基因组DNA为模板,以asl4561-F和asl4561-R为引物(见表1),Touch-down PCR扩增asl4561基因片段;以asl4562-F 和asl4562-R为引物(见表1),Touch-down PCR扩增asl4562基因序列片段.PCR反应程序为94℃ 4 min;94℃ 1 min,55 ℃(0.5 ℃↓)40 s,72 ℃ 40 s,15个循环;94 ℃ 1 min,47 ℃ 40 s,72 ℃ 40 s,20个循环;72 ℃ 7 min.引物中添加相应的酶切位点,PCR扩增产物纯化后与pMD18-T Vector载体连接,转化E.coilDH5α ,蓝白斑筛选阳性克隆酶切鉴定测序正确后保存质粒,分别命名为pMD18-T-4561和pMD18-T-4562.
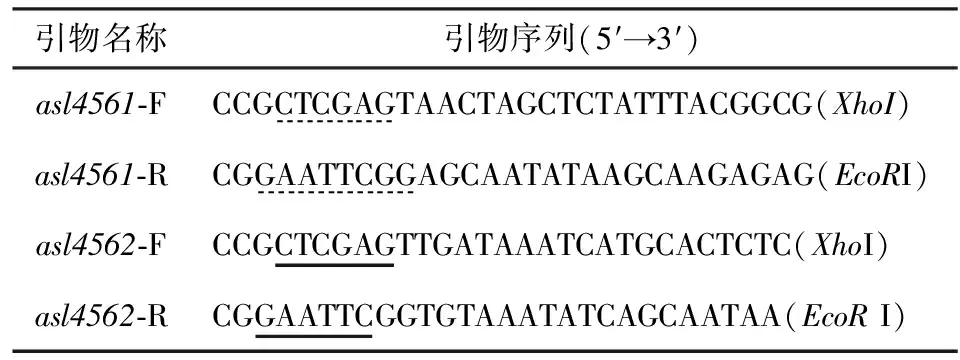
表1 PCR引物及序列
1.2.3 重组表达载体的构建
以XhoI和EcoRI分别双酶切质粒pMD18-T-4561和pET28a(+),电泳回收片段,连接转化E.coilBL21(DE3),以含50 μg/mL硫酸卡拉霉素的LB平板筛选转化子,重组质粒经XhoI/EcoRI双酶切鉴定,测序正确后命名为pET28a-4561.同理构建的含asl4562基因的重组质粒测序正确后命名为pET28a-4562.
1.2.4 目的蛋白的诱导表达
分别将含有重组质粒pET28a-4561和pET28a-4562的BL21接种于含50 μg/mL硫酸卡拉霉素的LB液体培养基中,37 ℃摇床培养约3 h至OD600=0.4~0.6后加入1 mmol/L的IPTG于28 ℃下摇床培养8h,诱导目的蛋白表达. 离心收集沉淀加入2×SDS凝胶缓冲液,沸水浴5 min,离心取上清用5 %的浓缩胶和15 %的分离胶进行SDS-PAGE电泳检测表达外源蛋白质的分子量.
1.2.5 pET28a-4561的蛋白表达条件的优化
1)最佳诱导时间试验. 含重组质粒pET28a-4561的BL21培养方法同上,当OD=0.4~0.6时,加入IPTG的终浓度为1.0 mmol/L、28℃振荡培养,分别在2, 4, 6, 8, 10h取样,离心收集沉淀,裂解后取上清SDS- PAGE电泳检测蛋白表达量.
2)最佳IPTG浓度试验.含重组质粒pET28a-4561的BL21培养方法同上,当OD600=0.4~0.6时,每管加入IPTG的终浓度分别为0.2, 0.4, 0.6, 0.8, 1.0 mmol/L,28 ℃诱导6 h后,离心收集菌体,处理后取上清SDS-PAGE 电泳检测蛋白表达情况.
2 结果
2.1 asl4561和 asl4562基因的克隆
通过Touch-down PCR扩增的asl4561和asl4562的基因片段产物经1.0%的琼脂糖凝胶电泳鉴定结果与预期大小(261bp和213bp)相符(见图1). 基于设计asl4561基因引物时两端外侧多取了24个碱基以及有酶切位点及保护碱基,因此检测结果约在250bp. pMD18-T-4561和pMD18-T-4562测序结果与NCBI所公布的asl4561和asl4562序列Blast比对,碱基完全相同说明成功克隆了两目的基因.
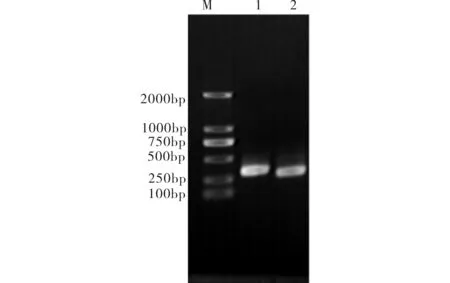
M)DL2000标准Mark;1)asl4561PCR产物;2)asl4562PCR产物
2.2 重组表达载体的构建
根据pET28a(+)多克隆位点选取EcoRI/XhoI为插入位点,将asl4561和asl4562 PCR产物与pET28a(+)经EcoR I/XhoI双酶切、纯化后的产物连接转化,挑取阳性克隆菌液PCR检测,并送测序发现asl4561和asl4562正确插入,插入片段分别为261bp和213bp,各自编码87个和71个氨基酸.
2.3 目的蛋白的诱导表达检测
含重组质粒pET28a-4561和pET28a-4562的BL21分别在1 mmol/L的IPTG、28 ℃下摇床培养8 h诱导表达,由SDS-PAGE电泳检测结果(见图2)可见,分别在15kD上方和10-15kD之间有过量表达的蛋白带.预测的asl4561和asl4562基因表达蛋白质分子量大小分别为10.46kD和8.36kD,又因pET28a(+)表达载体的组氨酸标签的密码子和相应的酶切位点,使产生的融合蛋白的分子量增加5.19kD,故产生的目的蛋白分子量分别为15.65kD和13.55kD,SDS-PAGE电泳检测结果大小与理论结果相符.
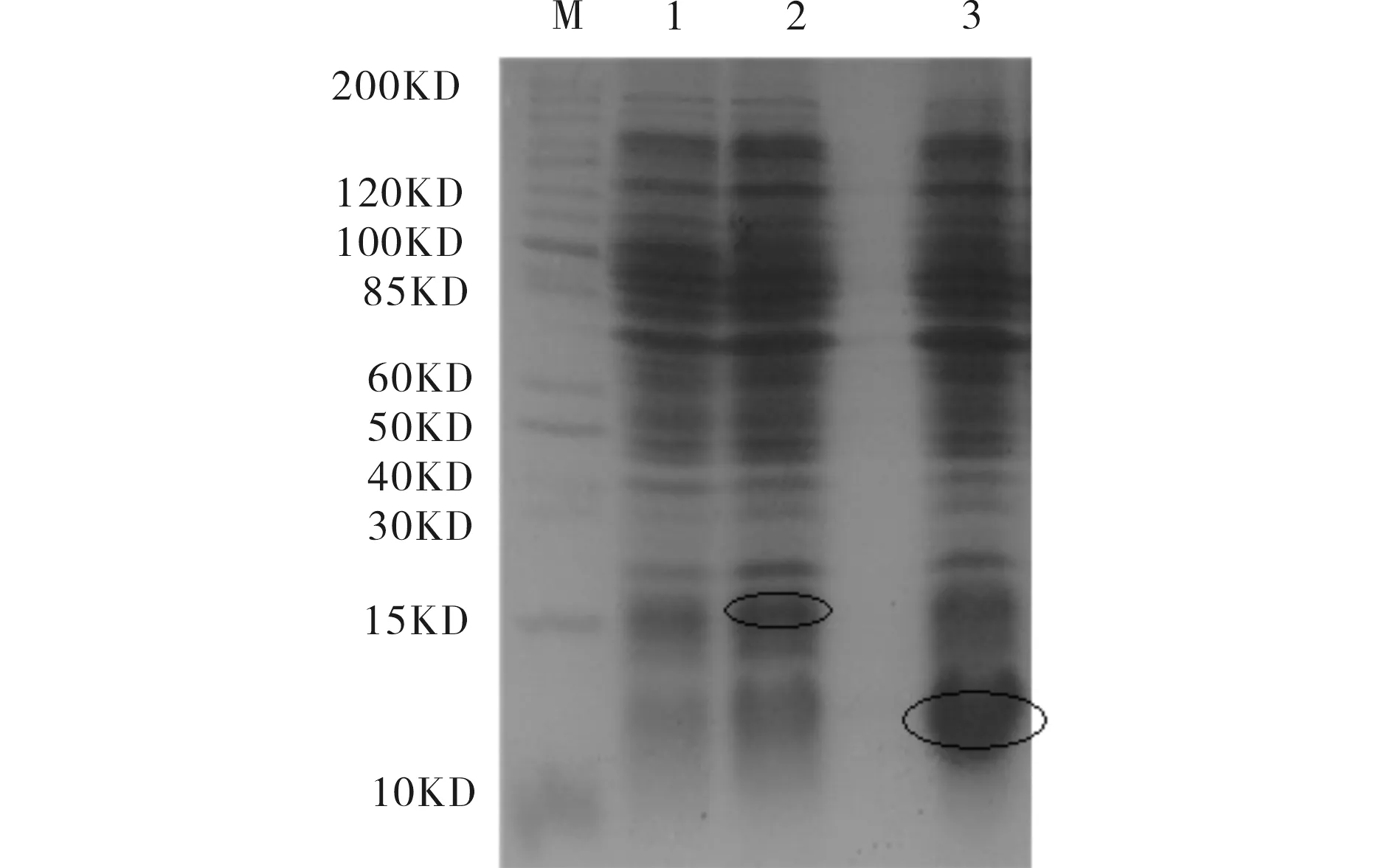
M)蛋白质分子量标准; 1)空载体诱导; 2)pET28a-4561诱导表达; 3)pET28a-4562诱导表达
2.4 蛋白表达条件优化结果
由图2蛋白检测图可见,asl4562基因在细菌裂解液上清中表达量比较大,基本可以满足后续的纯化;但asl4561基因表达蛋白量并非特别大,可能因为诱导条件不适合. 因此首先考虑在诱导剂IPTG浓度和诱导时间上对该蛋白的表达条件进行优化.
图3为不同诱导时间对蛋白表达量影响. 由图3可见,28 ℃、1.0 mmol/LIPTG诱导2 h取样已有目的蛋白表达,而6 h表达量增至最大,之后无明显增加,故可确定该蛋白最佳诱导时间为6 h.
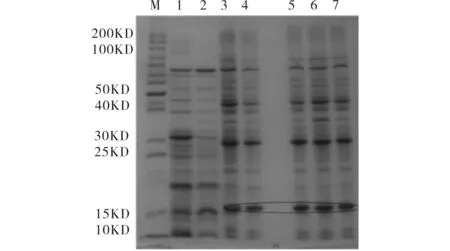
M)蛋白质分子量标准;1)空载体诱导后全菌; 2)未经诱导的pET28a-4561转化菌; 3~7)诱导时间依次为2, 4, 6, 8, 10 h的菌
图4为不同IPTG浓度对蛋白表达量影响. 由图4可见,在28 ℃下诱导6 h,当IPTG终浓度为0.4 mmol/L时,目的蛋白表达量最大,其余浓度的IPTG诱导表达量差异不太明显,均无IPTG为0.4 mmol/L时诱导表达量大,故确定最佳IPTG浓度为0.4 mmol/L.
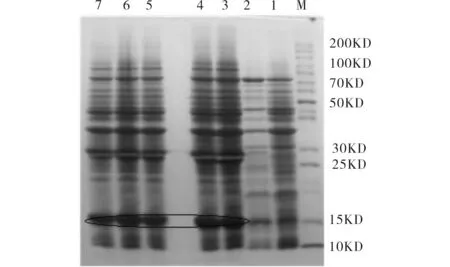
M)蛋白质分子量标准; 1)空载体诱导后全菌;2)未经诱导的pET28a-4561转化菌;3~7)IPTG浓度依次为0.2,0.4,0.6,0.8,1.0 mmol/L
3 分析与讨论
不同种属的细菌内TA系统的基因序列可能同源性较低或相差很大,但其遗传结构和功能却非常相似.本实验首先通过序列分析发现PCC7120染色体基因asl4561与大肠杆菌染色体上毒素基因relE有较高的同源性,并与其上游基因asl4562有4个碱基的重叠,与TA系统具有相同的遗传结构.通过对两基因编码产物的分析,推测它们在细胞生理条件下可通过静电引力形成复合物,有相互作用,初步推测asl4561和asl4562基因构成TA系统.
通过分子生物学方法构建的含asl4561基因的表达载体pET28a-4561和含asl4562基因的表达载体pET28a-4562,测序结果显示两基因完整的ORF均正确插入到pET28a(+)EcoR I/XhoI多克隆位点,无移码突变和点突变,表明重组表达载体构建成功.
蛋白检测结果显示两基因表达蛋白均存在于菌体裂解液上清中,说明此类蛋白为可溶性蛋白.asl4562基因在28 ℃、1 mmol/LIPTG 诱导8 h,蛋白表达量较大.在对重组质粒pET28a-4561表达条件的优化中发现28 ℃、0.4 mmol/LIPTG 诱导6 h,其蛋白表达量最大,说明低温、低浓度的IPTG诱导,可增加可溶性重组蛋白的产量[9].
近年研究证明TA系统作为原核细胞应对营养缺乏时的调控机制,是对细菌代谢调控的重要补充,对于设计新的药物解决细菌耐药性问题具有现实意义[10].但目前有关蓝细菌中TA系统的研究还不多,本实验结果为后续纯化并研究两蛋白之间的相互作用以及在它们鱼腥藻PCC7120体内的作用奠定基础.期望通过更深入的研究能为蓝细菌染色体上其他TA系统的研究奠定基础,由此入手可为除藻、解决水华问题提供新的思路.
[1]Gerdes K,Christensen S K,Lobner-Olesen A.Prokaryotic toxin-antitoxin stress response loci[J].Nature Reviews Microbiology,2005,3(5):371-382.
[2]Yamaguchi Y,Park J H,Inouye M.Toxin-antitoxin sys-
tems in bacteria and archaea[J].Annual Review of Genetics,2011,45:61-79.
[3]Lieven Buts,Jurij Lah,Minh-Hoa,et al.Toxin-antitoxin modules as bacterial metabolic stress managers[J].Trends in Biochemical Sciences,2005,30(12):672-679.
[4]Gronlund H,Gerdes K.Toxin-antitoxin syestems homo-
logous withrelBE ofEscherichiacoliplasmid P307 are ubiquitous in prokaryotes[J].J Mol Biol,1999,285(4):1401-1415.
[5]王晓蕾,赵龙旋,张俊杰.细菌毒素-抗毒素系统的研究进展[J].生物化学与生物物理进展,2008,35(9):991-997.
[6]Henriksen P.Estimating nodularin content of cyanoba-
cterial blooms from abundance of Nodularia spumigena and its characteristic pigments-a case study from the Baltic entrance area[J].Harmful Algae,2005(4):167-178.
[7]徐旭东,王业勤,黎尚豪.鱼腥藻-大肠杆菌CAT启动子探测质粒的构建[J].中国科学院研究生院学报,1993,10:203-209.
[8]萨姆布鲁克J, 拉塞尔D W.分子克隆实验指南[M]. 3版.黄培堂,译. 北京:科学出版社,2005.
[9]Ren Zengliang,Du Guocheng,Chen Jian,et al.Strategies for high-level expression of recombinant protein in Eschefichia coil[J].China Biotechnology,2007,27(9):103-109.
[10]季建军,邱景富,杨瑞馥,等. 细菌的细胞程序性死亡中毒素-抗毒素系统的研究进展[J].军事医学科学院院刊,2006,30(2):184-187.
