脂糖肽抗生素Telavancin的合成工艺改进*
2011-11-27梁玉华冯文化
梁玉华,冯文化
(中国医学科学院 北京协和医科大学 药物研究所,北京 100050 )
Telavancin{Nvan-2-(癸基氨基)乙基-29-[(N-膦酰基甲基)氨甲基]万古霉素(1)}是美国Theravance公司研发的新型脂糖肽抗生素。其注射剂于2009年被美国FDA批准上市,用于治疗成人复杂皮肤感染和皮肤结构感染,以及金黄色葡萄球菌引起的感染(包括甲氧西林敏感金黄色葡萄球菌和耐甲氧西林金黄色葡萄球菌)[1]。
在1的合成中,文献[2]方法以N-癸基氨基乙醇为原料,经Fmoc-Cl保护、Swerm氧化制得关键中间体N-(9-芴基甲氧羰基)-癸基氨基乙醛(8)。该方法存在DMSO沸点高难蒸除,需活化剂草酰氯,产生副产物甲基硫代亚甲基醚、卤代酮、二甲基硫醚等,操作繁琐,后处理麻烦,不易纯化,不宜工业化等缺陷。
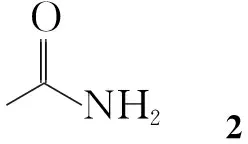
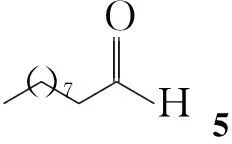
本文在文献[2]方法的基础上对8的合成工艺进行改进:癸醛(5)和氨基乙醛缩二甲醇缩合生成席夫碱,再经氰基硼氢化钠还原、Fmoc-Cl保护、酸水解制得8。再参照文献[2,3]方法将8与万古霉素盐酸盐(9, Chart 1)反应生成席夫碱,经氰基硼氰化钠还原、脱Fmoc保护制得Nvan-2-(癸基氨基)乙基万古霉素(10);10与氨甲基膦酸(4)进行曼尼希反应合成了目标化合物1(Scheme 1)。
合成8的改进路线具有提纯简单,后处理方便,三步收率(82.8%)较高等优点;在1的合成中,通过改变10与4的投料比,减少了副产物的生成,使单步(10→1)收率从27%[3]提高至48.7%,从而

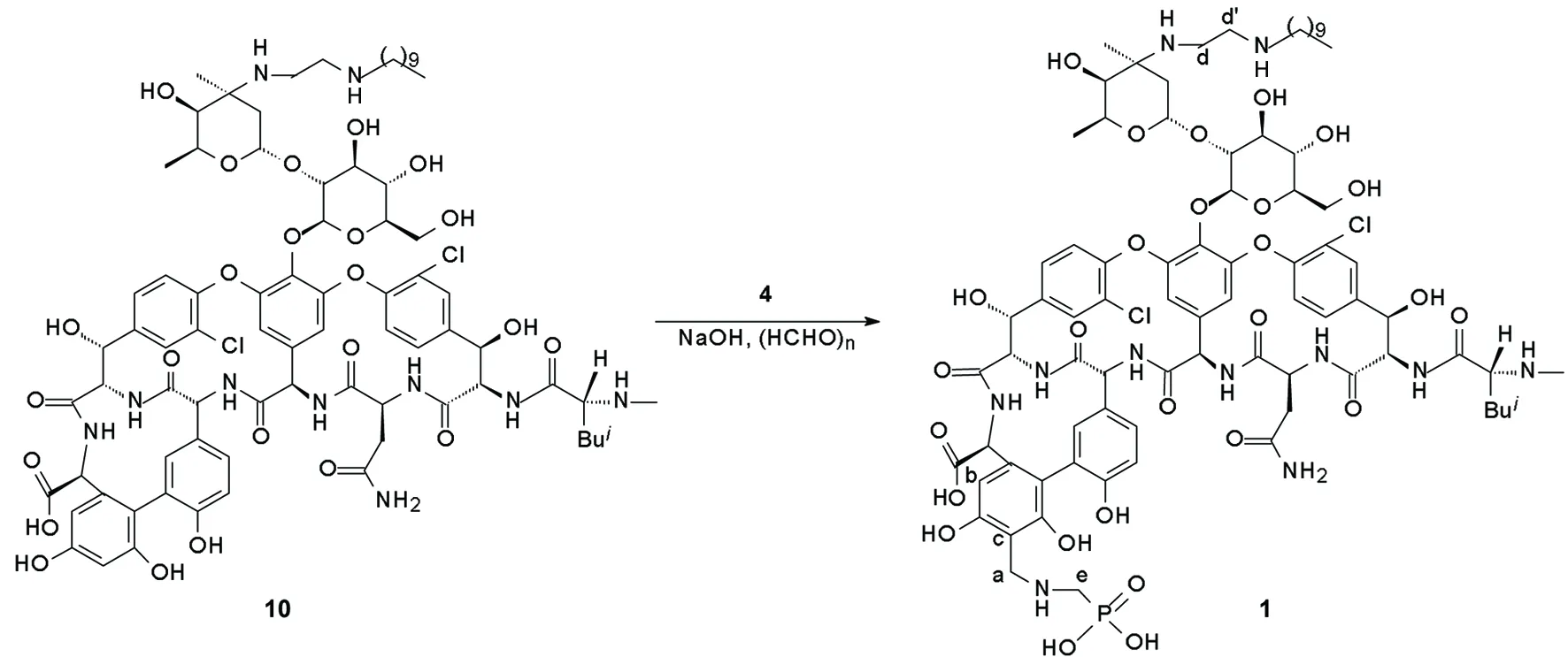
Scheme 1
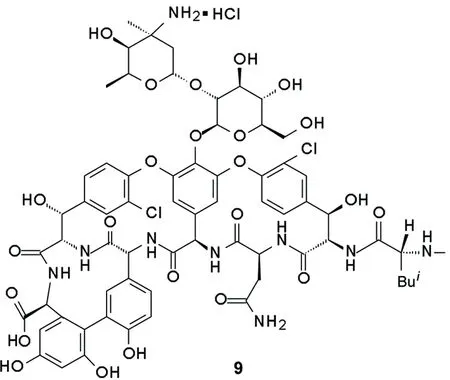
Chart 1
提高了1的总收率;对4的合成做了研究,通过乙酰胺(2),多聚甲醛及三氯化磷在冰醋酸中回流反应生成乙酰胺甲基膦酸(3); 3再经浓盐酸水解制得4,两步收率49.4%[4]。
1 实验部分
1.1 仪器与试剂
TRYT-3型熔点仪;Varain Mercury-600和Varain Mercury-400型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent LC/MSD TOF 型质谱仪;Alltech型高效液相色谱仪[HPLC,色谱柱:Kromasil C18柱(4.6 nm×150 mm, 5 μm);流动相:A=V(水)∶V(乙腈)∶V(TFA)=98∶2∶0.1%, B=V(水)∶V(乙腈)∶V(TFA)=10∶90∶0.1%;梯度洗脱:A∶B=95∶5~60∶40洗脱0 min~20 min,A∶B=60∶40洗脱20 min~40 min;流速:1.0 mL·min-1;检测波长:280 nm]。
乙腈,色谱纯;其余所用试剂均为化学纯;万古霉素盐酸盐,华北制药,纯度>93%。
1.2 合成
(1) 3的合成
在三口瓶中依次加入2 5.9 g(100 mmol),多聚甲醛3.3 g(110 mmol)和冰乙酸70 mL,搅拌下于100 ℃回流反应至白色固体完全溶解;冷却至室温,缓慢滴加三氯化磷16.5 g(120 mmol),滴毕,回流反应至终点(TLC检测)。减压蒸除冰醋酸后加入甲醇50 mL,抽滤,滤饼干燥得白色固体3 8.3 g,收率54.2%,m.p.184 ℃~186 ℃;1H NMR(D2O)δ: 3.42~3.51(d, 2H), 1.92(s, 3H); ESI-MSm/z: 154{[M+H]+}。
(2) 4的合成
在单口瓶中加入3 5.5 g(36 mmol)和8 mol·L-1盐酸50 mL,搅拌下于100 ℃回流反应至终点(TLC检测)。减压蒸除溶剂得棕色油状液体。加水100 mL和活性炭5 g,加热脱色反应2 h。抽滤,滤液减压旋蒸除水后加入甲醇80 mL,抽滤,滤饼干燥得白色固体4 3.66 g,收率91.5%,m.p.309 ℃~312 ℃;1H NMR(D2O)δ: 3.31~3.34(d,J=9.0 Hz, 2H);31P NMR(D2O): 13.6(s, 1P); ESI-MSm/z: 112{[M+H]+}。
(3)N-癸基氨基乙醛缩二甲醇盐酸盐(6)的合成
在反应瓶中加入5 4.68 g(30 mmol)和氨基乙醛缩二甲醇3.21 g(30.6 mmol)的甲醇(100 mL)溶液,搅拌下于室温反应12 h。冰浴冷却下分批加入氰基硼氢化钠2.83 g(45 mmol);于室温反应12 h(TLC检测)。蒸除溶剂,用3 mol·L-1盐酸调至酸性,用二氯甲烷(2×50 mL)萃取,合并有机层,用饱和食盐水洗涤两次,无水硫酸镁干燥,减压浓缩至干,加丙酮析晶,过滤,滤饼干燥得白色固体6 7.31 g,收率86.4%;1H NMRδ: 0.85~0.88(t, 3H), 1.24~1.31( m,4H),1.85~1.89(m, 2H), 3.04~3.06(m,4H),3.46(s, 6H),4.98~5.01(t, 1H)。
(4)N-(9-芴基甲氧羰基)-癸基氨基乙醛缩二甲醇(7)的合成
在反应瓶中加入6 2.82 g(10 mmol),DIPEA(N,N-二异丙基乙胺)2.71 g(21 mmol)及无水THF 80 mL,搅拌下于室温反应20 min;缓慢滴加Fmoc-Cl 2.58 g(10 mmol)的THF(30 mL)溶液,滴毕,反应至终点(TLC检测)。过滤,滤液减压蒸除THF后加入乙酸乙酯80 mL,依次用10%盐酸(2×50 mL)、饱和碳酸氢钠溶液(2×50 mL)和饱和氯化钠溶液(2×50 mL)洗涤,无水硫酸镁干燥,过滤,滤液减压浓缩至干得黄色油状液体7 4.61 g,收率98.5%;1H NMRδ: 0.81~0.81(t, 3H), 1.02~1.04(m, 1H), 1.17~1.36(m, 14H), 1.38~1.40(m, 1H), 2.98~3.07(m, 2H), 3.12(s, 3H), 3.15~3.22(m, 3H), 3.31(s, 3H), 4.15~4.18(m, 1H), 4.37~4.39(m, 1H), 4.42~4.43(d,J=6.0 Hz, 1H), 4.52~4.53(d,J=5.2 Hz, 1H), 7.18~7.69(m, 8H)。
(5) 8的合成
在反应瓶中加入7 4.0 g的THF(50 mL)溶液,浓盐酸10 mL,搅拌下于室温反应至终点(TLC检测)。减压蒸除THF,加入乙酸乙酯100 mL,依次用饱和碳酸氢钠溶液(2×100 mL)、饱和氯化钠溶液(2×100 mL))洗涤,无水硫酸镁干燥,过滤,滤液减压浓缩至干得黄色油状液体8 3.51 g,收率97.3%;1H NMRδ: 0.87~0.88(t, 3H), 1.10~1.43(m, 14H), 1.69~1.76(m, 2H), 3.10~3.14(m, 2H), 3.68~3.73(m, 2H), 4.23~4.25(m,1H), 4.52~4.55(m, 2H), 7.18~7.69(m, 8H)。
(6)10的合成
氮气保护,在反应瓶中加入9 4.0 g(2.69 mmol), 8 1.47 g(3.5 mmol)及DIPEA 1.04 g(8.07 mmol)及DMF 40 mL,搅拌使其完全溶解。加入氰基硼基化钠0.34 g,于室温反应至终点(HPLC检测)。加入甲醇10 mL和三氟乙酸(TFA)0.92 g,于室温反应1 h。减压蒸除甲醇,加水300 mL,析出大量白色沉淀,减压抽滤,滤饼用DMF溶解后加入二乙胺0.6 g,反应5 h;加入丙酮300 mL,析出大量白色固体,减压抽滤,滤饼用乙酸乙酯(2×100 mL)洗涤,干燥得白色固体10 3.27 g,收率74.5%,纯度83.60%(HPLC,下同); ESI-MSm/z: 817.323 9{[M+2H]2+}。
(7) 1的合成
在反应瓶中加入10 2.0 g(1.225 mmol )和4 271 mg(2.45 mmol)的乙腈(12 mL)溶液,搅拌10 min;加水8 mL,用3 mol·L-1NaOH溶液调至pH 9~10,于室温反应15 min。加入多聚甲醛38.59 mg(1.29 mmol),反应至终点(HPLC检测)。用3 mol·L-1盐酸调至pH 2~3,加入丙酮150 mL,析出白色固体,减压过滤,滤饼用乙酸乙酯(2×100 mL)洗涤后经反相硅胶柱层析[洗脱剂:V(乙腈)∶V(水)=2∶8]纯化得白色固体1 1.05 g,产率48.7%,纯度91.8%(91.3%[5]);1H NMR(DMSO)δ: 0.81~0.86(m, 6H), 0.93~0.94(d, 3H), 1.05~1.07(d, 3H), 1.17~1.41(br, 16H), 1.43(s, 3H), 1.62~1.65(m, 4H), 1.86~1.93(m, 2H), 2.07(s, 2H), 2.49~2.60(m, 2H), 2.72~2.95(m, 8H), 3.18~3.66(m, 19H), 3.98(m, 1H), 4.14(br, 1H), 4.24(s, 1H), 4.40(s, 1H), 4.65(br, 1H), 4.84(br, 1H), 5.08~5.29(m, 7H), 5.61~5.71(m, 2H), 6.03~6.10(br, 1H), 6.49(s, 1H), 6.69~6.78(m, 3H), 6.95(br, 1H), 7.05~7.58(m, 7H), 7.83(s, 1H), 8.62(br, 4H), 8.97(s, 1H), 9.36(s, 1H), 9.51(s, 1H); ESI-MSm/z: 878.808 4{[M+2H]2+}。
2 结果与讨论
与文献[6]1H NMR数据相比发现,1在6.29少了间苯二酚上一个氢的特征信号(c-H),而在6.49有b-H的特征信号,在5.08~5.29,3.18~3.66存在a-H和e-H信号,表明万古霉素只在c-位上进行了曼尼希反应。在0.81~0.86,1.17~1.41上分别为Nvan-位上的葵基取代基的甲基峰和亚甲基峰。而d,d′-H的化学位移在2.72~2.95。
在由10合成1的过程中,文献[3]方法用r[n(10)∶n(4)=1∶6,此时1会进一步与4反应生成二曼尼希副产物;若r=1∶1时,则原料转化不完全;本文通过工艺优化,发现r=1∶2时,只得到单曼尼希产物(1),原料转化最佳,产率由27%[3]提高至48.7%。
1,9和10的极性相近,使用普通的薄层色谱难以分离,需使用高效液相监测反应。文献[3,5]方法用的是C14色谱柱,本文通过优化液相条件,使用C18色谱柱即可将1,9和10分离,方便操作。
[1] 尚新艳,阮丽军,许激扬,等.第二代糖肽类抗生素的研究进展[J].中国抗生素杂志,2007,32(5):263-312.
[2] Lee J.Process for preparing glycopeptide derivatives[P].WO 03 018 607,2003.
[3] Michael R L, Stacy M A, Bettina B,etal.Hydrophobic bancomycin derivatives with improved ADME properties:Discovery of telacancin(TD-6424)[J].The Journal of Antibaiotics,2004,57(5):326-336.
[4] Heinsohn G E.Process for the preparation of phosphorous acid/carboxylic acid mixed anhydrides and anhydrous phosphorous acid[P].WO 9 513 285,1995.
[5] Liu Y W, Lee J.Hydrochloride salts of a glycopetide phosphonate derivative[P].US 7 531 623,2005.
[6] Dudley H W, John R K.Structural and mode of action studies on the antibiotic vancomycin.Evidence from 270-MHz proton magnetic resonance[J].JACS,1976,99(8):2768-2774.
