肥胖与免疫炎症
2011-11-26王旭方综述刘志红审校
王旭方 综述 刘志红 审校
随着生活方式的改变,肥胖发病率在世界范围内呈上升趋势。传统观点认为肥胖是一种代谢性疾病,能引起胰岛素抵抗,进而成为2型糖尿病、心血管疾病和脂质代谢紊乱的高危因素。但随着肥胖患者脂肪组织肿瘤坏死因子 α(tumor necrosis factor-α,TNF-α)等炎症因子的发现,肥胖作为一种慢性炎症性疾病的观点已得到共识。肥胖的一些并发症,例如胰岛素抵抗,也被认为与慢性炎症有关。
近年来关于肥胖的研究越来越多的集中于免疫炎症领域。在发现了脂肪组织巨噬细胞募集的现象后,很多学者就引起巨噬细胞募集的因子、肥胖组织巨噬细胞分型以及其他种类免疫细胞的功能做了广泛而深入的研究。因此,认识免疫炎症与代谢的关系可能为临床治疗肥胖提供一个方向。
肥胖、炎症与胰岛素抵抗
糖代谢中胰岛素的主要作用靶点是肝脏、骨骼肌和脂肪组织,它们对胰岛素及其他种类激素的反应决定了血糖、血脂等代谢产物的水平。生理状况下血糖水平主要由两方面因素决定,一是肝糖输出,胰岛素能抑制此过程;二是肌肉组织对葡萄糖的摄取,胰岛素能激活此过程。两者失衡则引起糖代谢异常。另外,胰岛素能促进脂肪组织摄取脂肪酸并以三酰甘油的形式储存,同时避免三酰甘油的脂解作用。
肥胖状态下,这三种胰岛素作用靶细胞均可使促炎通路活化,从而引起胰岛素抵抗,使肝糖输出增加、肌肉对葡萄糖利用减少以及游离脂肪酸从脂肪组织中释放(图1)[1]。一项肥胖患者脂肪组织及血液样本的基因表达研究发现,有关炎症、免疫等基因的表达也显著上调[2]。南京军区南京总医院全军肾脏病研究所对肥胖相关性肾病患者肾小球基因表达谱的分析表明,除与脂代谢和胰岛素受体相关的基因外,一些炎性因子如 TNF-α、白细胞介素6(interleukin-6,IL-6)、干扰素 γ(interferon-γ,IFN-γ)的表达亦明显升高[3]。这从基因层面上证明肥胖不仅仅是一种代谢异常,同时也是一种炎症性疾病。肥胖患者脂肪细胞和巨噬细胞可以分泌很多细胞因子和趋化因子如单核细胞趋化因子配体2(chemokine c-c motif ligand 2,CCL-2)、IL-6、IL-1β和TNF-α等,这些因子介导炎症反应。肥胖动物模型脂肪组织和外周血TNF-α水平都升高,其升高与胰岛素抵抗呈正相关,药物抑制TNF-α可减轻肥胖引起的胰岛素抵抗[4]。进一步证实肥胖、免疫炎症与胰岛素抵抗之间存在一定的关系。
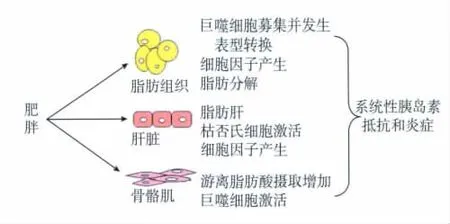
图1 肥胖个体脂肪组织、肝脏和骨骼肌改变引起胰岛素抵抗和炎症[1]
脂肪组织在炎症反应中的作用
脂肪组织是肥胖患者炎症反应中的一个重要始动环节。它不仅储存多余能量,且通过自分泌或旁分泌的形式释放脂肪酸及多肽类,包括激素、细胞因子和趋化因子。脂肪组织中包含多种细胞,如脂肪细胞、前脂肪细胞、免疫细胞(巨噬细胞、淋巴细胞等)和内皮细胞。脂肪细胞可特异性的分泌增强胰岛素敏感性的脂肪因子,如瘦素和脂联素,也可分泌引起胰岛素抵抗的蛋白,如抵抗素和视黄醇结合蛋白4[5]。因此,在特定病理生理状态下,这些脂肪因子对于系统性胰岛素抵抗起重要作用。
脂肪组织巨噬细胞(adipose tissue macrophage,ATM)的发现为肥胖与炎症反应的关系提供了一个研究方向。ATM含量与肥胖程度成正相关,正常人ATM在脂肪组织所有细胞中的比例小于10%,而在肥胖状态下可高达40%[6]。最初认为ATM的功能是吞噬凋亡的脂肪细胞,进一步研究发现ATM是脂肪组织促炎因子的主要来源,活化的巨噬细胞可分泌多种趋化因子,引起越来越多的巨噬细胞向脂肪组织浸润,形成正反馈,加重慢性炎症过程。同时,巨噬细胞也能通过旁分泌作用于胰岛素靶细胞,激活核因子κB抑制蛋白激酶β(inhibitor ofκB kinase β,IKKβ)和 c-Jun氮末端激酶(Jun N-terminal kinase,JNK)等丝氨酸激酶,引起脂肪细胞的胰岛素抵抗[7,8]。近期也有研究表明巨噬细胞可作用于前脂肪细胞[9]。体外实验发现巨噬细胞分泌的促炎因子能使前脂肪细胞表型发生改变,并分泌一些纤维成分,促进自身的迁移和增生,进而引起脂肪细胞数量增多,肥胖加重。肝脏和骨骼肌巨噬细胞活化
肥胖患者肝脏胰岛素抵抗主要与炎症介质表达上调和肝细胞内脂肪聚积相关。肝脏巨噬细胞主要是肝窦中的枯否氏细胞。关于枯否氏细胞的研究比ATM少,但它也能分泌促炎因子并且对肝细胞胰岛素抵抗起着重要作用。敲除枯否氏细胞的炎症反应基因不能阻止脂肪肝的发生,但可减轻肝细胞胰岛素抵抗[10]。
高热量饮食也可引起骨骼肌组织巨噬细胞的募集。和脂肪组织一样,骨骼肌巨噬细胞主要是促炎的M1型,这些细胞主要存在于肌肉组织间的脂肪中[10]。这说明巨噬细胞可能通过旁分泌途径引起骨骼肌胰岛素抵抗,但是迄今尚未有直接证据来证实。骨骼肌巨噬细胞浸润数量明显少于脂肪组织和肝脏。
巨噬细胞促炎通路
巨噬细胞内一个重要的促炎通路是IKKβ系统。静息状态下核因子 κB(nuclear factor-κB,NF-κB)抑制蛋白(inhibitor of NF-κB,IκB)与 NF-κB形成一个复合物,将NF-κB限制在细胞质中。肥胖状态下IKKβ激活,使IκB磷酸化,与NF-κB解离并降解。NF-κB进入细胞核与相应DNA结合,激活炎症基因,引起单核细胞趋化蛋白1(monocyte chemoattractant protein-1,MCP-1)和TNF-α等炎症因子表达增加。另一个研究比较多的细胞内促炎通路是JNK1系统。在这个通路里,外源性促炎信号引起JNK的磷酸化和激活,使c-Jun的N末端磷酸化,进而c-Jun二聚体变成异源性c-Jun/c-Fos二聚体,从而激活炎症基因。在高热量饮食下,巨噬细胞IKKβ(或JNK1)敲除小鼠与同等饮食条件下野生型小鼠肥胖程度相同,但糖耐量异常和高胰岛素血症明显改善,骨骼肌、肝脏和脂肪组织对胰岛素的敏感性也得到提高[8,11]。同时,这些组织中巨噬细胞数量和炎症因子水平也显著下降。其重要意义在于,巨噬细胞IKKβ(或JNK1)敲除的小鼠同样可以获得肥胖和脂肪肝,但它们并未产生胰岛素抵抗,证明肥胖不一定存在胰岛素抵抗,无炎症作用情况下单纯肥胖不会引起胰岛素抵抗发生。
巨噬细胞向脂肪组织募集
早期研究认为肥胖患者脂肪组织中坏死或凋亡的脂肪细胞可招募巨噬细胞。进一步研究发现脂肪组织招募了外周血一部分单核细胞。肥胖个体循环中细胞间黏附分子1(intercellular adhesion molecule-1,ICAM-1)表达增加,使单核细胞与血管内皮黏附,并且浸润到脂肪组织中[12]。脂肪细胞能够分泌集落刺激因子1(colony stimulating factor-1,CSF-1),促使单核细胞分化为成熟的巨噬细胞[13]。Westcott等[14]发现CD301在7/4hi单核细胞向炎症组织的迁移中起了重要作用,敲除CD301的动物模型,循环中7/4hi单核细胞水平下降,并且不会出现胰岛素抵抗和ATM募集。
趋化因子及其受体(C-C chemokine receptor 2,CCR-2)在人体皮下和内脏脂肪组织中高表达,它们大多源于脂肪组织间质血管结构,也有少量由脂肪细胞分泌。其中,MCP-1及其受体CCR-2是迄今为止研究最广泛的。大多研究认为MCP-1过表达可以引起ATM募集[14],而缺乏MCP-1或CCR-2可以减少ATM募集,改善胰岛素抵抗[15]。但是它们的确切作用目前尚存在争议,也有报道 MCP-1或CCR-2缺乏并不能减少ATM的数量[16]。人体内,CCL-5也被证明与内脏脂肪组织炎症基因表达以及巨噬细胞浸润呈正相关[17]。体内研究发现CCL-5可以激活循环中单核细胞向脂肪组织血管内皮的黏附和迁移,并能阻止巨噬细胞凋亡,保留其清除脂类的能力,因此在巨噬细胞生存中起着一定作用。
除趋化因子外,一些炎症介质也能通过促进趋化因子的表达而引起巨噬细胞募集。Mamane等[18]证明缺乏补体C3a受体(C3aR)的小鼠,其ATM浸润明显减少,并且可以抵抗饮食诱导的肥胖、胰岛素抵抗和脂肪肝。这证明补体系统也在ATM募集中起着一定作用。
同样,糖基化终产物、游离脂肪酸等营养物质也可以引起脂肪组织巨噬细胞募集。它们通过活性氧(reactive oxygen species,ROS)产生、丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)、NF-κB和过氧化物酶增生物激活受体γ(peroxisome proliferator-activated receptorγ,PPARγ)的激活等机制来调控MCP-1等趋化蛋白的表达[19],进而引起炎症反应。
巨噬细胞分型及影响因素
巨噬细胞是脂肪组织中最早发现的炎性细胞,通过经典活化和替代活化两条途径产生两个不同表型[20]。经典途径中,IFN-γ和脂多糖(lipopolysaccharides,LPS)激活 M1型巨噬细胞,分泌促炎因子(如TNF-α、IL-6),产生ROS。替代途径中,IL-4和IL-13激活M2型巨噬细胞,产生IL-10和转化生长因子β(transforming growth factor-β,TGF-β),抑制由M1型巨噬细胞介导的炎症反应过程。同时M2型巨噬细胞在组织修复等非免疫炎症过程中也发挥作用。M2型巨噬细胞又可分为许多亚型,其中 M2b型不仅可以表达抑炎因子(如IL-10),同时也可以表达促炎因子(如TNF-α),且促炎因子的表达水平甚至高于M1型巨噬细胞,而M2a型和M2c型主要表达抑炎因子[21]。M1型巨噬细胞可以表达CD11c,而M2型不表达。在高热量饮食诱发的肥胖小鼠中可观察到巨噬细胞从M2型向M1型转化,伴随着CD11c表达增强。CD11c对于脂肪组织T细胞的浸润和激活以及胰岛素抵抗起着重要作用。CD11c敲除小鼠脂肪组织炎症反应减轻,T细胞数量下降,脂肪组织对胰岛素的敏感性得以改善[22]。
PPARγ对于巨噬细胞表型转化起着重要的调节作用。巨噬细胞PPARγ敲除的小鼠体内炎症通路激活,在正常饮食条件下也可出现糖耐量异常和胰岛素抵抗[23]。而激活PPARγ能使ATM增多,但增多的巨噬细胞以 M2型为主[24],但也有报道PPARγ活化后可通过抑制MCP-1和 CCR-2引起ATM数量减少[25]。另外,PPARγ激活可诱导 M2b型巨噬细胞向M2a型转化[24]。事实上,替代途径中M2型巨噬细胞的成熟必须要有PPARγ表达[20]。巨噬细胞PPARγ表达对于正常机体的胰岛素敏感性起着重要作用,缺乏PPARγ可引起糖耐量减退以及骨骼肌和肝脏的胰岛素抵抗[23]。PPARγ也能通过促进脂肪酸β氧化来降低游离脂肪酸水平,从而减轻炎症反应[20]。PPARδ在ATM中也起着类似作用,体外培养去除PPARδ的巨噬细胞很难向M2型转化,从而引起炎症反应[26]。在体内,PPARδ主要影响肝脏枯否氏细胞表型。因此,在PPARγ主要影响ATM表型的同时,可能PPARδ的主要作用靶点在肝脏。
很多研究发现饱和脂肪酸是促炎的,不饱和脂肪酸的促炎作用比较微弱,而ω3不饱和脂肪酸可以抑制炎症反应。进一步研究发现Toll样受体4(Toll-like receptor 4,TLR4)在饱和脂肪酸促炎过程中起着重要作用。肥胖个体中,高浓度游离脂肪酸能够激活巨噬细胞TLR4,通过调控NF-κB、干扰素调控因子(interferon regulatory factor,IRF)等转录因子活性诱发大量促炎基因表达,从而使巨噬细胞分泌 TNF-α、IL-6 和 IL-1β等促炎因子[27]。正常小鼠静脉输注大量脂肪酸后可以出现炎症反应和胰岛素抵抗,而TLR4敲除小鼠则不会出现此现象[28]。这些实验证明TLR4可以识别游离脂肪酸,促进巨噬细胞向M1型转化,引起炎症反应。
T淋巴细胞与脂肪组织炎症
巨噬细胞是脂肪组织中发现最早、研究最多的细胞,但最近的报道也陆续发现其他种类白细胞的浸润情况(图2)。人脂肪组织淋巴细胞可以表达CCR-6,它作为脂肪细胞分泌的CCL-20受体,可能是淋巴细胞与脂肪细胞相互作用的一个媒介[29]。
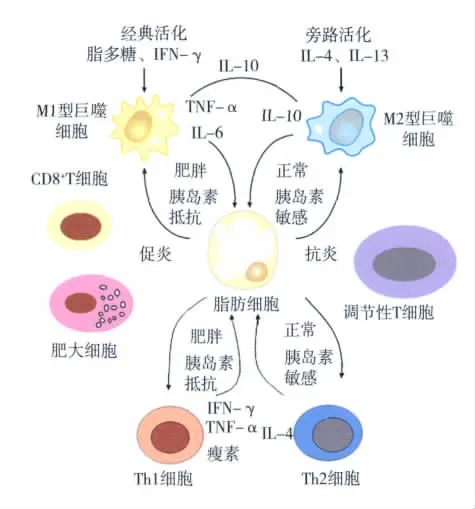
图2 肥胖个体脂肪组织炎症细胞类型及其作用[30-32,34]
Nishimura等[30]证明脂肪组织中CD8+T细胞对于巨噬细胞浸润起着重要作用。首先,他们发现肥胖小鼠附睾组织CD8+T细胞数量明显增多,并且早于巨噬细胞出现;然后他们证实敲除CD8+T细胞的小鼠即使给予高热量饮食也未出现大量巨噬细胞浸润,并且这些小鼠的胰岛素抵抗也得以改善,而过继转移CD8+T细胞后脂肪组织炎症加重;最后,体外实验表明肥胖个体脂肪组织的CD8+T细胞可使正常脂肪组织中的巨噬细胞分化为分泌TNF-α的亚型。这个研究很清晰的证实了CD8+T细胞在肥胖脂肪组织中对于巨噬细胞的募集、分化和激活作用。
CD4+T细胞在肥胖个体脂肪组织中也发挥着相应的作用[31]。CD4+T细胞有分泌IFN-γ的 Th1细胞和分泌IL-4、IL-13的Th2细胞两种亚型。在正常脂肪组织中,两者比例相当;而肥胖状态下,Th1细胞数量明显上升,伴随Th2细胞数量下降,Th1/Th2比例失衡最终可导致脂肪组织炎症发生、M1型巨噬细胞浸润及胰岛素抵抗。同一项研究表明,缺乏T淋巴细胞和B淋巴细胞的Rag1和Rag2基因敲除小鼠,其肥胖和胰岛素抵抗程度比野生型小鼠更重。这可能与内脏脂肪组织中自然杀伤细胞和巨噬细胞浸润有关,而通过基因重组获得CD4+T细胞并缺乏CD8+T细胞的小鼠,它们的体重明显下降,并且ATM浸润数量也显著减少。这些研究显示CD4+Th2细胞可以减轻内脏脂肪的炎症反应。
调节性T细胞(Treg)在免疫应答中起调控作用,它可以通过调控其他种类的淋巴细胞和固有免疫细胞的数量来避免不恰当的免疫应答。肥胖小鼠脂肪组织Treg的数量是明显降低的。Feuerer等[32]证明,缺乏Treg的小鼠,其肝脏及脂肪组织对胰岛素的敏感性均下降,而炎症因子水平明显上升。激活Treg则能引起小鼠血糖水平的下降,同时刺激IL-10的分泌。另外,转录水平显示脂肪组织Treg有其独特的功能,体现在促使白细胞外移的基因表达增强。这个层面进一步揭示了Treg在调控脂肪组织炎症中不可替代的作用。
作为一种固有免疫淋巴细胞,自然杀伤 T(natural killer T cells,NKT)细胞近期才被发现与脂肪组织的炎症相关[33]。它是一种固有的细胞毒性T细胞,可识别脂类抗原。研究认为肥胖小鼠脂肪组织中NKT细胞数量是增加的,基因敲除引起NKT细胞缺乏的小鼠不会获得高三酰甘油血症,也不会引起ATM浸润。反之,NKT细胞激活能引起糖耐量减退和ATM浸润。因此,有理由认为脂肪组织中NKT细胞对于其他免疫细胞的聚集起着一定作用,并且脂类抗原的存在可促进免疫细胞向脂肪组织浸润。肥大细胞与巨噬细胞的关系
传统观点认为肥大细胞主要介导过敏反应,但新近研究表明,肥大细胞也参与了糖尿病和肥胖患者的炎症反应。Liu等[34]证明在食物诱导肥胖小鼠脂肪组织中,肥大细胞的浸润早于巨噬细胞。而同样饮食条件下肥大细胞敲除小鼠或者应用肥大细胞稳定剂的小鼠,其体重、ATM数量及炎性因子表达均明显下降,提示肥大细胞可能参与了ATM的募集。另外,肥大细胞表达的IL-6和IFN-γ可增加脂肪组织一些蛋白水解酶的表达,这些酶分解抗血管生成的分子,从而促进脂肪组织血管发生。因此,肥大细胞诱导的血管发生为脂肪组织中更多白细胞的浸润提供了一个途径。
抗炎治疗及意义
既然炎症对于肥胖个体胰岛素抵抗如此重要,药物阻断炎症反应能起到治疗效果吗?体内实验证实中和fa/fa糖尿病大鼠的TNF-α可改善胰岛素抵抗[4];但临床试验证实TNF-α阻滞剂依那西普并不能改善代谢综合征患者的胰岛素抵抗[35]。一个可能的原因是TNF-α是通过旁分泌起作用的,循环中TNF-α浓度要比组织中低1~2个数量级。而依那西普作为一个大分子TNF-α阻滞剂,即使在很高浓度下也不能渗入细胞间隙,因此,尽管循环中TNF-α已经被有效抑制了,但组织中TNF-α并未得到抑制,它仍然可以发挥胰岛素抵抗的效应。
大剂量水杨酸治疗可抑制2型糖尿病患者的IKKβ,改善糖耐量,减轻胰岛素抵抗[36]。同样,对于血糖正常而存在高胰岛素血症的患者使用双水杨酯可降低空腹、餐后血糖及游离脂肪酸,同时脂联素水平得以恢复。这些研究证明将IKKβ及炎症作为治疗靶点可以改善胰岛素抵抗。
最初人们认为噻唑烷二酮类(thiazolidinedion,TZDs)可直接作用于脂肪细胞和骨骼肌细胞使之增强胰岛素敏感性,但是最近研究发现,敲除骨髓PPARγ的小鼠,尽管体重在正常范围,但仍可以存在高胰岛素血症,并且它们对于TZDs的治疗效果并不理想[23],这说明TZDs可能是通过PPARγ来起作用的。迄今无研究能表明TZDs可以促进巨噬细胞分泌改善胰岛素敏感性的因子,但它无疑可以抑制促炎因子的产生。这类药物的作用机制仍需进一步研究。
南京军区南京总医院肾脏病研究所报道中药雷公藤甲素能抑制 TNF-α、IL-6、IL-1β 和 IFN-γ等多种因子生成,并且抑制NF-κB活性,下调CD8+T细胞数量,同时不影响胸腺功能和正常免疫反应,体内实验也表明它可显著降低db/db小鼠肾组织慢性低度炎症和氧化应激[37],证明雷公藤甲素在治疗肥胖引起的慢性炎症方面有其独特的优势。
小结:慢性炎症在肥胖患者病情进展中起着重要作用,既往研究就脂肪组织炎症细胞浸润以及炎症与胰岛素抵抗的关系做了较为详尽的阐述,但仍有许多未知领域等待探索。另外,抗炎治疗对于改善这类患者的胰岛素抵抗有较好的疗效,但抗炎药物的种类选择及其作用机制仍有待进一步研究。
1 Schenk S,Saberi M,Olefsky JM.Insulin sensitivity:modulation by nutrients and inflammation.J Clin Invest,2008,118(9):2992-3002.
2 Emilsson V,Thorleifsson G,Zhang B,et al.Genetics of gene expression and its effect on disease.Nature,2008,452(7186):423-428.
3 Wu Y,Liu Z,Xiang Z,et al.Obesity-related glomerulopathy:insights from gene expression profiles of the glomeruli derived from renal biopsy samples.Endocrinology,2006,147(1):44 -50.
4 Hotamisligil GS,Shargill NS,Spiegelman BM.Adipose expression of tumor necrosis factor-alpha:direct role in obesity-linked insulin resistance.Science,1993,259(5091):87 -91.
5 Scherer PE.Adipose tissue:from lipid storage compartment to endocrine organ.Diabetes,2006,55(6):1537 -1545.
6 Weisberg SP,Hunter D,Huber R,et al.CCR2 modulates inflammatory and metabolic effects of high-fat feeding.J Clin Invest,2006,116(1):115-124.
7 Hirosumi J,Tuncman G,Chang L,et al.A central role for JNK in obesity and insulin resistance.Nature,2002,420(6913):333 -336.
8 Vallabhapurapu S,Karin M.Regulation and function of NF-kappaB transcription factors in the immune system.Annu Rev Immunol,2009,27:693-733.
9 Keophiphath M,Achard V,Henegar C,et al.Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes.Mol Endocrinol,2009,23(1):11 - 24.
10 Arkan MC,Hevener AL,Greten FR,et al.IKK-beta links inflammation to obesity-induced insulin resistance.Nat Med,2005,11(2):191-198.
11 Solinas G,Vilcu C,Neels JG,et al.JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity.Cell Metab,2007,6(5):386 -397.
12 Sengenes C,Miranville A,Lolmede K,et al.The role of endothelial cells in inflamed adipose tissue.J Intern Med,2007,262(4):415-421.
13 Ferrante AW,Jr.Obesity-induced inflammation:a metabolic dialogue in the language of inflammation.J Intern Med,2007,262(4):408-414.
14 Westcott DJ,Delproposto JB,Geletka LM,et al.MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity.J Exp Med,2009,206(13):3143 -3156.
15 Kanda H,Tateya S,Tamori Y,et al.MCP-1 contributes to macrophage infiltration into adipose tissue,insulin resistance,and hepatic steatosis in obesity.JClin Invest,2006,116(6):1494 -1505.
16 Kirk EA,Sagawa ZK,McDonald TO,et al.Monocyte chemoattractant protein deficiency fails to restrain macrophage infiltration into adipose tissue[corrected].Diabetes,2008,57(5):1254 -1261.
17 Keophiphath M,Rouault C,Divoux A,et al.CCL5 promotes macrophage recruitment and survival in human adipose tissue.Arterioscler Thromb Vasc Biol,2010,30(1):39 - 45.
18 Mamane Y,Chung Chan C,Lavallee G,et al.The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation.Diabetes,2009,58(9):2006 -2017.
19 Yeop Han C,Kargi AY,Omer M,et al.Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes:dissociation of adipocyte hypertrophy from inflammation.Diabetes,2010,59(2):386-396.
20 Odegaard JI,Ricardo-Gonzalez RR,Goforth MH,et al.Macrophagespecific PPARgamma controls alternative activation and improves insulin resistance.Nature,2007,447(7148):1116 -1120.
21 Mosser DM,Edwards JP.Exploring the full spectrum of macrophage activation.Nat Rev Immunol,2008,8(12):958 -969.
22 Wu H,Perrard XD,Wang Q,et al.CD11c expression in adipose tissue and blood and its role in diet-induced obesity.Arterioscler Thromb Vasc Biol,2010,30(2):186 -192.
23 Hevener AL,Olefsky JM,Reichart D,et al.Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones.JClin Invest,2007,117(6):1658-1669.
24 Lefevre L,Gales A,Olagnier D,et al.PPARgamma ligands switched high fat diet-induced macrophage M2b polarization toward M2a thereby improving intestinal Candida elimination.PLoS One,2010,5(9):e12828.
25 Guri AJ,Hontecillas R,Ferrer G,et al.Loss of PPAR gamma in immune cells impairs the ability of abscisic acid to improve insulin sensitivity by suppressing monocyte chemoattractant protein-1 expression and macrophage infiltration into white adipose tissue.J Nutr Biochem,2008,19(4):216 -228.
26 Kang K,Reilly SM,Karabacak V,et al.Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity.Cell Metab,2008,7(6):485 -495.
27 Nau GJ,Richmond JF,Schlesinger A,et al.Human macrophage activation programs induced by bacterial pathogens.Proc Natl Acad Sci U SA,2002,99(3):1503 -1508.
28 Saberi M,Woods NB,de Luca C,et al.Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice.Cell Metab,2009,10(5):419-429.
29 Duffaut C,Zakaroff-Girard A,Bourlier V,et al.Interplay between human adipocytes and T lymphocytes in obesity:CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators.Arterioscler Thromb Vasc Biol,2009,29(10):1608 -1614.
30 Nishimura S,Manabe I,Nagasaki M,et al.CD8+effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity.Nat Med,2009,15(8):914 -920.
31 Winer S,Chan Y,Paltser G,et al.Normalization of obesity-associated insulin resistance through immunotherapy.Nat Med,2009,15(8):921-929.
32 Feuerer M,Herrero L,Cipolletta D,et al.Lean,but not obese,fat is enriched for a unique population of regulatory T cells that affect metabolic parameters.Nat Med,2009,15(8):930 -939.
33 Ohmura K,Ishimori N,Ohmura Y,et al.Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice.Arterioscler Thromb Vasc Biol,2010,30(2):193-199.
34 Liu J,Divoux A,Sun J,et al.Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice.Nat Med,2009,15(8):940 -945.
35 Bernstein LE,Berry J,Kim S,et al.Effects of etanercept in patients with the metabolic syndrome.Arch Intern Med,2006,166(8):902-908.
36 Goldfine AB,Silver R,Aldhahi W,et al.Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes.Clin Transl Sci,2008,1(1):36 - 43.
37 Gao Q,Shen W,Qin W,et al.Treatment of db/db diabetic mice with triptolide:a novel therapy for diabetic nephropathy.Nephrol Dial Transplant,2010,25(11):3539 -3547.
