高效液相色谱/串联质谱法测定水产品中环丙沙星的不确定度
2011-10-14颜斌曹军陈勇杨瑞章徐邦兴王太全张丹高玲
颜斌 曹军 陈勇 杨瑞章 徐邦兴 王太全 张丹 高玲
(盐城出入境检验检疫局 江苏盐城 224002)
高效液相色谱/串联质谱法测定水产品中环丙沙星的不确定度
颜斌 曹军 陈勇 杨瑞章 徐邦兴 王太全 张丹 高玲
(盐城出入境检验检疫局 江苏盐城 224002)
对高效液相色谱/串联质谱法测定水产品中环丙沙星含量的不确定度来源进行分析与量化。测试样品中环丙沙星含量测量结果的扩展不确定度为1.21μg/kg(k=2)。
高效液相色谱/串联质谱法;水产品;环丙沙星;测量不确定度
1 前言
目前高效液相色谱/串联质谱法已广泛用于水产品中喹诺酮类兽药(环丙沙星)的定性定量检测,为了使其符合CNAS-CL07:2006《测量不确定度评估和报告通用要求》[1],且在测量数据的处理、测量结果的表达和测量结果质量评定等方面与国际接轨,本文根据CNAS-GL06:2006《化学分析中不确定度的评估指南》[2],对高效液相色谱/串联质谱法测定水产品中环丙沙星的测量结果不确定度进行了测量和评定,为科学评定测量结果质量提供依据。
2 材料与方法
2.1 材料
2.1.1 仪器
Thermo Finnigan TSQ Quantum Access液相色谱-高分辨串联四极杆质谱联用仪,配有电喷雾电离源。
2.1.2 试剂
环丙沙星标准品:购自Sigma-Aldrich公司,纯度≥98%。甲醇:Merck,色谱纯;乙腈:南京化学试剂厂,分析纯;无水硫酸钠:南京化学试剂厂,分析纯;正己烷:南京化学试剂厂,分析纯。
2.2 方法
准确称取粉碎均质鮰鱼肌肉组织样5.00g(±0.01g)于50mL离心管中,加入约10g无水硫酸钠,涡旋混匀;加入15 mL乙腈提取液,涡旋混匀2min;3500 rpm离心10min,取上层清液,再用10mL提取液提取2次,涡旋混匀2min;3500 rpm离心10min,合并上层清液,于50℃水浴下旋转蒸发至干。准确加入1.00mL甲醇水混和溶液(5:95),涡旋后再加入0.5 mL正己烷涡旋,转移入1.5mL离心管,15000rmp离心5min,取下层溶液过0.45μm的滤膜到进样瓶中,上仪器测定。
3 不确定度评定过程
3.1 数学模型

其中:ω为样品中环丙沙星的质量浓度(μg/kg);
c为待测样品溶液中环丙沙星的质量浓度(μg/L);
v为待测溶液的定容体积(mL);
m为称取样品的质量(g);
frec为回收率校正因子。
3.2 不确定度分量的主要来源
3.2.1 校准过程引入的不确定度
包括采用最小二乘法拟合标准工作曲线求得试样浓度过程中所引入的不确定度、标准储备液稀释成标准溶液过程所引入的不确定度。而标准储备液稀释成标准溶液过程中的不确定度由标准储备液浓度的不确定度、移液管和容量瓶体积的不确定度、校准和使用温度不同导致的不确定度组成。
3.2.2 样品质量引入的不确定度
由天平的最大允许误差构成。
3.2.3 体积引入的不确定度
由定容体积和进样体积的不确定度组成。
3.2.4 制样过程引入的不确定度
测试样品的制备过程非常复杂,需经过均质、萃取、过滤、净化、淋洗、浓缩等步骤,每一步操作都会引入不确定度,要确定每一步骤对测量结果不确定度的贡献相当困难,可采用检测方法确认中的有关数据如回收率等,对制样过程引入的不确定度进行评定。
3.2.5 测试过程随机效应导致的不确定度
包括样品的均匀性和代表性、天平的重复性、体积刻度充满的重复性、进样的重复性和仪器积分面积重复性等因素引入的不确定度。
4 不确定度分量的评定
4.1 校准过程引入的不确定度(属B类不确定度)
4.1.1 标准物质引入的不确定度
4.1.1.1 标准品不确定度
环丙沙星标准储品纯度≥98%,纯度误差为±0.5%,按矩形分布处理,其标准不确定度为:u(Cs)相对标准不确定度为:urel(Cs)=0.29%/98%=0.0030
4.1.1.2 标准溶液配制过程引入的不确定度
(1)100μg/mL标准储备溶液的配制
称取10.00mg环丙沙星标准储品100 mL容量瓶(A级)中,用乙腈定容。使用天平规定允差为0.05mg,根据GB12806-1991《实验室玻璃仪器单标线容量瓶》[3]规定:A级单标线100mL容量瓶的容量允许差为±0.10mL,取矩形分布,则引入的标准不确定度为:

100μg/mL标准储备溶液的配制过程的相对标准不确定度为:

(2)1μg/mL标准溶液的配制
用1 mL移液器将1 mL100μg/mL标准储备溶液完全转移至100 mL容量瓶(A级)中,用乙腈定容。根据GB12806-1991规定:A级单标线100mL容量瓶的容量允许差为±0.10mL,JJG 646-2006《移液器检定规程》[4]规定:1mL移液器吸取1mL溶液容量允许差为1%,按矩形分布处理,标准不确定度为:

1μg/mL标准溶液的配制过程的相对标准不确定度为:

(3)按上述步骤计算200μg/L、100μg/L、50μg/L、20μg/L、10μg/L和5μg/L标准溶液配制过程的相对标准不确定度分别为0.0105、0.0129、0.0183、0.0129、0.0129、0.0129。计算时取50μg/L浓度标准溶液的不确定度值0.0183(最大不确定度值)。
标准溶液的配制过程的相对标准不确定度为:4.1.1.3 温度效应

温度:容量瓶在20℃校准,本测试在25℃的条件下进行。因为溶液的体积膨胀系数远大于玻璃的膨胀系数,因此只考虑前者即可。乙腈体积膨胀系数为1.37×10-3℃-1,产生的体积变化为100mL×5℃×1.37×10-3℃-1=0.685mL,取矩形分布,则溶液温度对体积的影响所引入的标准不确定度为:u(Vtemp)=0.685/3=0.3955(mL);相对不确定度为:urel(Vtemp)=0.3955/100=0.0040。
4.1.1.4 标准物质引入的相对不确定度
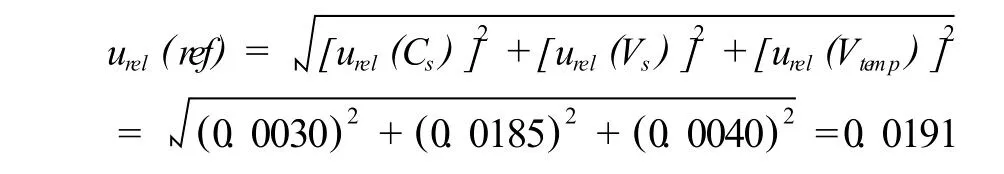
4.1.2 采用最小二乘法拟合标准工作曲线求得试样浓度过程中所引入的不确定度
配制环丙沙星的6个不同浓度标准溶液(200μg/L、100μg/L、50μg/L、20μg/L、10μg/L和5μg/L),分别用LC-MS/MS测试3次,得到相应的峰面积A,采用最小二乘法拟合,得到标准工作曲线的线性回归方程Y=aX+b(b为截距,a为斜率)和方程的线性相关系数,见表1。

表1 环丙沙星峰面积、线性回归方程、线性相关系数
对样品溶液进行3次测定,根据峰面积用标准工作曲线的线性回归方程求得的平均浓度(C0)为110.40μg/L,则由最小二乘法拟合标准工作曲线所引入的标准不确定度为:

其中:a=71136;b=32053,试验3次P=3,n=6×3=18
Scc为标准溶液浓度残差的平方和
由最小二乘法拟合标准工作曲线引入的相对标准不确定度为:

4.1.3 标准过程引入的不确定度

4.2 样品质量引入的不确定度(属B类不确定度)
天平的说明书给出的最大允许误差0.01g,按矩形分布处理,标准不确定度和相对标准不确定度分别为:u(m)0.0058/5.0=0.0012
4.3 体积引入的不确定度(属B类不确定度)
4.3.1 由定容体积引入的不确定度
样品经过萃取、净化后用1mL移液器移取1mL甲醇水混和溶液(5:95)溶解,根据JJG 646-2006中要求,按矩形分布处理,相对标准不确定度及和相对标准不确定度分别为:
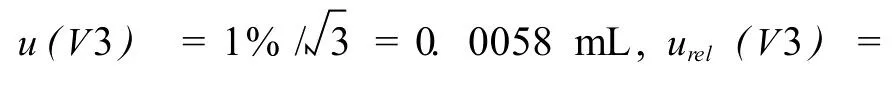
4.3.2 由进样体积的引入的不确定度
测试中仪器配置的微量注射器体积为25μL,进样量为25μL,根据JJG700-1999附录A《微量注射器的标准》[5],微量注射器体积的相对标准偏差为1%,按矩形分布处理,则进样体积引入的相对标准不确定度为:
4.3.3 由体积引入的相对不确定度

4.4 前处理过程引入的不确定度(属A类不确定度)
制样过程产生的不确定度采用100μg/L(与所测定样品浓度水平相近)5次样品添加回收率的相关数据进行计算。3次回收率测定结果为:91.5%、93.5%、95.6%、94.2%、92.7%,平均回收率为R=93.5%,标准偏差为S=1.54%,标准不确定度和相对标准不确定度分别为:
u(R)=S=0.0154
urel(R)=u(R)/R=0.0165
4.5 测试过程随机效应导致的不确定度(属A类不确定度)
按照SN/T 1751.2-2007《动物源性食品中16种喹诺酮类药物残留量检测方法》[6]要求,测定鮰鱼样品中环丙沙星残留量,结果分别为:22.19μg/kg,22.01μg/kg,22.04μg/kg,平均值ω—=22.08μg/kg,标准偏差s(ω—)=0.0964μg/kg,按矩形分布处理,则测试过程随机效应导致的标准不确定度和相对标准不确定度分别为:u(ω—)=s(ω—)=0.0964,urel(ω—)=u(ω—)/ω—=0.0044
4.6 合成不确定度
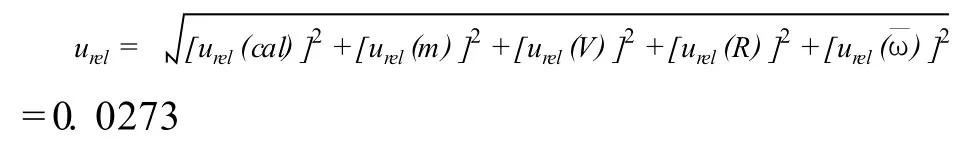
4.7 扩展不确定度
扩展不确定度可由合成标准不确定度乘以包含因子,在95%置信水平下,k=2时,试样中环丙沙星含量测定的相对扩展不确定度为:
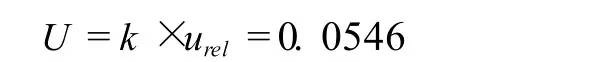

4.8 测量结果的报告
测试鮰鱼样品中环丙沙星测量结果:
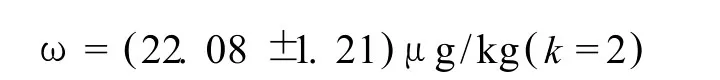
[1] CNAS CL07:2006测量不确定度评估和报告通用要求[S].
[2] CNAS GL06:2006化学分析中不确定度的评估指南[S].
[3] GB12806-1991实验室玻璃仪器单标线容量瓶[S].
[4] JJG 646-2006移液器检定规程[S].
[5] JJG700-1999气相色谱仪检定规程[S].
[6] SN/T 1751.2-2007动物源性食品中16种喹诺酮类药物残留量检测方法[S].
Uncertainty of Measurement of Ciprofloxac in Residues in Aquatic Products by HPLC-MS/MS
Yan Bin,Cao Jun,Chen Yong,Yang Ruizhang,Xu Bangxing,Wang Taiquan,Zhang Dan,Gao Ling
(Yancheng Entry-Exit Inspection and Quarantine Bureau,Yancheng,Jiangsu,224002)
The uncertainty of measurement in determination of ciprofloxacin in aquatic products by HPLC-MS/MS was evaluated.The main sources of uncertainty resulted from the determination process were discussed and calculated.The expanded uncertainty of ciprofloxacin determination of was 1.21μg/kg(k=2).
HPLC-MS/MS;Aquatic Products;Ciprofloxacin;Uncertainty of measurement
O657.63
国家质检总局科研计划项目(2010IK186)
