基于不同标准化假设分析癌基因组甲基化谱
2011-08-13吴瑞红程立新李朋飞王明月王晨光
王 栋 吴瑞红 程立新 李朋飞 王明月 李 宾 王晨光 郭 政*
1(电子科技大学生命学院,成都 610054)
2(哈尔滨医科大学生物信息科学与技术学院,哈尔滨 150086)
引言
异常的DNA甲基化与癌的发生发展密切相关[1-2],因此在全基因组范围内识别癌相关的差异甲基化基因可以分析疾病发生的分子机制,发现用于疾病诊断与药物靶设计的生物学标志[3]。然而,利用甲基化芯片检测的高通量数据往往受诸如试剂与检测批次、不同操作者等多种因素的影响[4-5]。通常认为,这些技术变异可能降低检测生物信号的统计效能[6]。为了排除这些技术变异的影响,针对基因表达谱数据处理开发的标准化方法(如分位数标准化[7]和中值标准化[8]等)常被应用于处理甲基化谱数据。这些标准化方法将所有样本的信号值都标准化到相同的分布,或者使之具有相同的中值,为此通常需要假设在疾病样本中差异表达的基因很少,并且差异高表达和低表达的基因数目大致相等[5]。然而,最近的研究显示,在癌表达谱中包含大量上调表达的基因。采用这些传统假设的标准化方法,会失查很多与癌相关的上调差异表达基因,并且发现很多假的下调差异表达基因[9],因此需要采用不依赖于如前所述假设的数据标准化方法,如最小变化集(least variant set,LVS)等[10]。通过分析低通量癌基因组甲基化谱数据,有研究者认为癌基因组呈现广泛的低甲基化状态[11-12],因此在癌样本和正常样本中的甲基化信号归一化的标准化方法可能会扭曲真实的生物学信号。对此问题,研究者并未达成共识。一些研究人员在分析DNA甲基化谱数据时选择不进行标准化处理[13-14],而大部分研究者仍然采用常规的标准化方法对甲基化谱数据进行预处理[15-16]。不同的处理方法可能对筛选差异甲基化基因等后续分析有重要的影响,有必要进行全面的评估。
在本研究中,通过分析4种癌型的各2套甲基化谱数据,发现癌样本与正常样本的原始信号值(beta值)的中值无显著差异。基于原始信号值和分位数标准化后信号值筛选出来的差异甲基化基因交叠比例很高,并且改变方向(高甲基化或低甲基化)的高度一致。基于分位数标准化后信号值可以筛选出更多的差异甲基化基因,而且这些基因的改变方向与其对应原始信号的改变方向高度一致,提示采用分位数标准化可以提高识别差异甲基化基因的统计效能,且并未对甲基化数据造成严重的偏倚。此外,由同一种癌型的不同数据集得到的差异甲基化基因,其改变方向呈现高度的一致性,提示利用甲基化谱可以可靠地发现癌症中甲基化的改变方向。
1 材料与方法
1.1 数据
本研究所用数据来自高通量基因表达数据库(Gene Expression Omnibus,GEO)[17]和癌症基因组图谱数据库(The Cancer Genome Atlas,TCGA)[18],如表1所示。所有的数据都产生自 Illumina HumanMethylation27 BeadChip平台。去除性染色体上的位点后,共包括26486个位点,涉及13890个基因。对于来自癌症基因组图谱中的数据集,使用了该数据库中定义的 Level2数据,包含探针甲基化(M)与非甲基化(U)信号强度值、探针与基因的对应关系。甲基化程度Beta值的计算采用Beta=M/(U+M+100)[14]。
由于甲基化谱中的数据可能受到不同批次和不同实验室等因素的影响[4],本研究采取如下的标准选取数据集:选择包含不少于10对(癌及癌旁正常组织的配对)样本的数据集,每个数据集中的全部癌症与正常配对样本来自同一个批次及同一个实验室,对每个癌型选取两个具有最大样本量的数据集。数据集采用如下的命名规则:癌型(配对样本数)。通过筛选,最终采用了结肠癌、肺癌、肾癌和胃癌的甲基化数据 (见表1)。
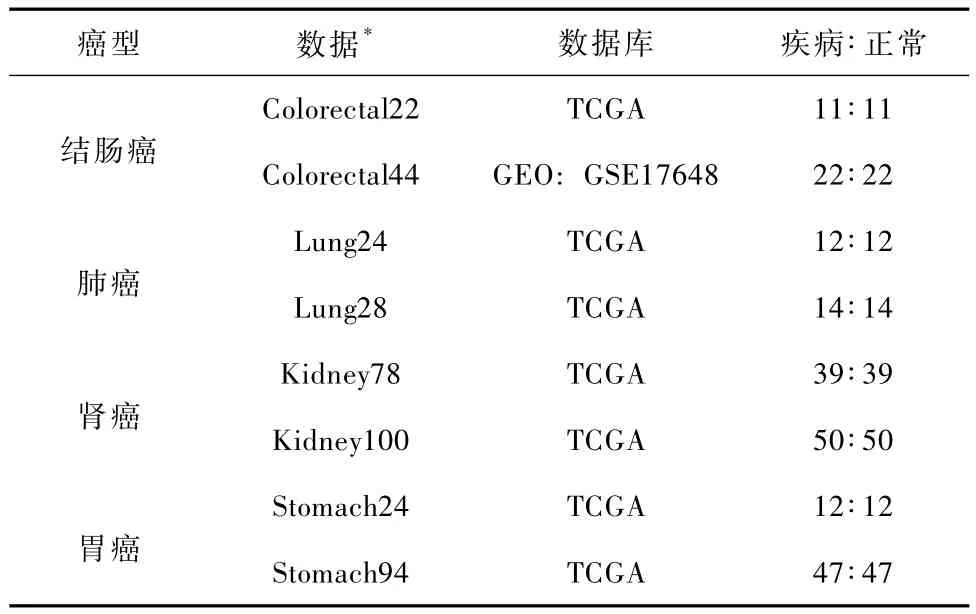
表1 4种癌型的甲基化数据Tab.1 The methylation datasets for four cancer types
1.2 分位数标准化算法
分位数标准化算法的目的,是使所有样本中探针强度的分布具有一致的分位数[7]。假设有 M张芯片(样本),每张芯片上有 N个探针,则构造一个N×M维的探针强度矩阵E,通过将矩阵的每个列向量按大小排秩,得到矩阵E1。然后,对E1的每行取均值,得到参考向量。最后,将参考向量中的探针表达值对应到E中每个列向量的原有秩次上,这样所有样本中的探针强度就保持了一致的分布[7]。如图1所示,原始数据中不同样本间整体甲基化水平波动较大,通过分位数标准化,可以使不同样本间高通量甲基化数据信号更加一致。通常认为,如此标准化后,可以消除技术变异对高通量甲基化数据所带来的影响,使得数据具有较好的可比性。
有研究显示,分位数标准化算法能够获得与其他复杂的标准化算法相同甚至更好的效果[19];又由于其计算简单,因而被广泛采用。值得注意的是,由于在高通量芯片数据中复杂的技术噪声难以准确估计[19],对标准化算法的评价通常只是经验性的,而并不能够根据其数学原理做简单的解释。
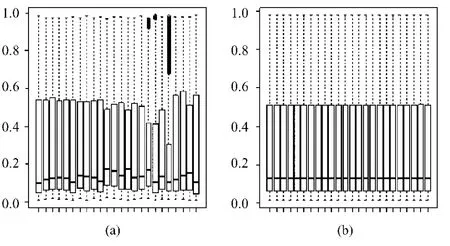
图1 甲基化数据。(a)标准化前;(b)标准化后Fig.1 The methylation datasets before.(a)and after;(b)normalization
1.3 预处理与差异甲基化基因的筛选方法
每套数据都采用两种处理方法:一是对样本进行分位数标准化,使beta值在所有的样本间都具有相同的分布[7];二是不对 beta值进行标准化处理。对于有多个甲基化检测位点的基因,保留所有检测位点具有一致的甲基化改变方向的基因。对每种癌型的两套数据,只将其中筛选出来的基因交集作为后续分析。
采用配对 t检验识别差异甲基化基因,并用Benjamini-Hochberg方法来控制多重检验的错误发现率[20]。
1.4 一致性分析
对于得到的两个差异基因列表,采用基因重合比例(percentage of overlapping genes,POG)指标,评价两个癌基因列表的一致性[21]。如果长度为 L1的列表1与长度为L2的列表2有k个交叠的基因,那么从列表1到列表2方向的基因重合比例得分为k/L1,即POG12=k/L1,从列表2到列表1方向的基因重合比例得分为k/L2,即POG21=k/L2。同时,还计算了列表1中的差异甲基化基因在列表2中的方向改变一致的比例(P1),列表2中找到的差异甲基化基因在列表1中方向改变一致的比例(P2)。采用伯努利(Bernoulli)检验,判断一致性得分显著性水平。
对采用同一种方法在同一种癌型的不同数据集中选择的差异甲基化基因列表,采用同样的方法来评价其改变方向的一致性得分及其显著性。
2 结果
2.1 标准化对甲基化数据信号的影响
在每套数据集中,首先对每个样本取其所有检测位点的原始 beta值的中值,然后利用配对秩和(wilcoxon ranksum)检验分析正常样本和癌症样本的beta值的中值有无显著差别。在这8套数据中,均未发现显著差异(P>0.05)。控制错误发现率(false discovery rate,FDR)为 0.01,采用配对 t检验,在每套数据中筛选差异甲基化基因;再使用基因重合比例指标,评估采用原始数据和分位数标准化数据筛选出的差异甲基化基因的一致性。结果显示(见表 2),在各数据集中,POG12的指标为 80% ~96%,即有80% ~96%的由采用原始数据筛选出来的差异甲基化基因包括在由分位数标准化数据筛选出的差异甲基化基因中。例如,在 kidney100数据中,采用原始数据筛选到了3274个差异甲基化基因,其中有2992(89%)个包括在由分位数标准化数据筛选出的3451个差异甲基化基因中,并且这些共同筛选到的2992个基因在标准化前后的甲基化改变方向相同。但是,在各数据集的原始数据中筛选出来的差异甲基化基因中,有4% ~20%的基因没有在分位数标准化数据中被筛选出来。因此,虽然正常和癌样本的beta值的中值差别不显著,但是数据标准化过程仍然可能会影响部分基因的显著性判断,在减少技术变异的同时也可能增加一定的偏倚[22]。
POG21指标为72% ~88%,反映在各数据集中有12~28%的采用分位数标准化数据筛选出来的差异甲基化基因,没有在原始数据中筛选出来。为了分析在一套数据中由分位数标准化数据单独筛选出来的差异甲基化基因是否确实为反映癌与正常样本差异的生物学有效信号,分析这些基因在关于同种癌型另外一套独立非数据集中原始信号的改变方向。假设如果这些被判断为差异甲基化的基因其实是与疾病状态无关的随机错误,则它们的差异改变方向将不会在独立实验中被保留下来。分析结果显示:对各癌型,在一套数据中由分位数标准化数据单独筛选出来的差异甲基化基因,在另外一套数据中原始信号改变方向的一致性为80% ~99%(见表3),均显著高于随机期望值50%(伯努利检验,P<0.001)。例如,在 kidney100数据中,采用分位数标准化数据单独找到529个差异甲基化基因,其中的470(89%)个基因的改变方向在kidney78数据的原始数据中得到验证,显著高于随机期望(伯努利检验,P<0.001)(见表3)。因此,大部分在各套数据中采用分位数标准化数据筛选出的差异甲基化,确实为反映癌与正常样本差异的生物学有效信号。
在各套数据中,采用原始数据筛选出的差异甲基化基因数目,都少于采用分位数标准化数据筛选出的差异甲基化基因,提示在原始数据中筛选差异基因的统计效能可能较低。例如,TUSC2(tumor suppressor candidate 2)和TUSC3(tumor suppressor candidate 3)在两套肾癌的分位数标准化数据中,均被判断为差异高甲基化基因,而采用原始数据分析均未能够将这两个基因判断为差异甲基化基因。已经有研究证实,TUSC2和 TUSC3均为抑癌基因,它们的高表达可以诱导细胞凋亡,而高甲基化状态可以抑制这两个基因的表达从而促进癌症的发展[23-24]。再如,WIF1(WNT inhibitory factor 1)在两套肾癌的分位数标准化数据中,均被判断为差异高甲基化基因;而采用原始数据分析均未将这个基因判断为差异甲基化基因。已有研究证实,WIF1的高表达可以抑制细胞生长和促进细胞凋亡,发挥抑癌基因的作用;而在高甲基化状态时,则不能发挥抑癌作用[25-26]。
采用原始数据和分位数标准化数据分别筛选出差异甲基化基因,其改变方向具有高度的一致性,P1和P2指标为99% ~100%(见表2)。例如,在胃癌的两套数据中,采用原始数据和分位数标准化数据,分别筛选出的差异表达基因的改变方向完全一致。
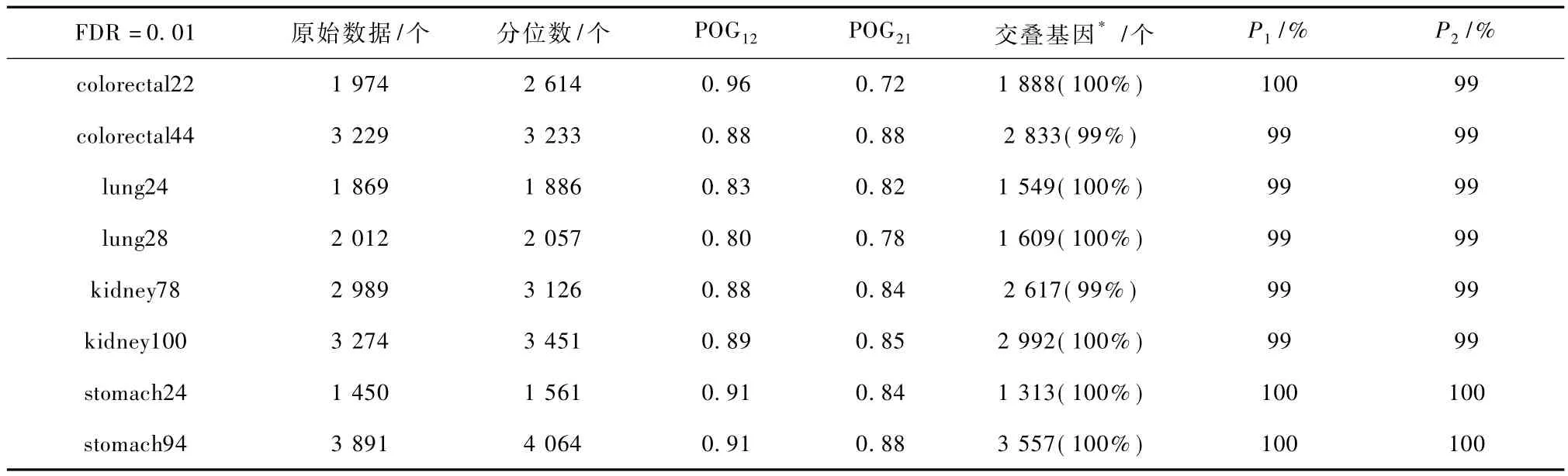
表2 在标准化与非标准化数据中筛选的差异甲基化基因改变方向的一致性Tab.2 The consistency of methylation states of DM genes separately selected from the normalized and non-normalized data

表3 由分位数标准化额外筛选出的差异基因在独立数据中甲基化状态的一致性Tab.3 The consistency of methylation states of DM genes solely selected by the Quantile method in one dataset with their methylation states in another independent data
但是,值得注意的是,在结肠癌、肺癌和肾癌中,在经过分位数标准化数据筛选出的差异甲基化基因中,约有1%基因的甲基化状态与原始数据中信号的改变方向相反。因此,数据标准化过程也可能会出现一定的过标准化现象,从而产生错误的结果。
2.2 差异甲基化基因识别的可重复性
由于大部分由采用原始数据筛选出来的差异甲基化基因包括在由分位数标准化数据筛选出的差异甲基化基因的列表中,所以只评价采用分位数标准化数据处理识别差异甲基化基因的可重复性。对每种癌型的两套数据分别采用分位数标准化处理后,所获得的差异甲基化基因的甲基化改变方向高度一致(见表4)。例如,在肾癌两套数据中,都被识别为差异甲基化的2876个基因的甲基化改变方向全部相同(伯努利检验,P<0.001)。另外,95%以上的仅在一套数据中找到的差异甲基化基因在另一套数据中有相同的改变方向,反映这些基因在另一套数据中存在着有效的生物信号。例如,有575个基因仅在kidney100数据中被判断为差异甲基化基因,但是其中的567个(99%)基因在kidney78数据中具有相同的甲基化改变方向,显著高于随机期望(伯努利检验,P <0.001)。
因此,在关于一种癌型的不同的高通量数据集中,识别的差异甲基化基因有高度的可重复性。这个结果也提示,在小样本数据中找到的差异甲基化基因相对少的原因可能是:由于严格的控制多重检验假阳性的统计过程,降低了识别差异甲基化基因的统计效能[21,27]。
3 讨论与结论
数据标准化,是利用基因甲基化谱识别差异甲基化基因等后续分析的基础。在分析低通量癌基因组甲基化数据时,一些研究者发现癌基因组呈现广泛的低甲基化现象[12],因此认为传统的标准化方法可能会扭曲真实的甲基化信号[28]。然而,低通量甲基化谱数据并不一定能够准确反映全基因组的甲基化特点,可能会产生偏倚。通过分析关于4种癌型的各两套高通量全基因组的甲基化谱数据,发现在癌症和正常样本中,探针信号的中值分布并没有显著的差别,提示癌症和正常样本的基因组整体甲基化状态并无显著差别。另外,由于存在检测变异(噪声),采用原始数据筛选差异甲基化基因的统计效能可能会降低,从而会导致许多差异甲基化基因不能被筛选出来,而经过分位数标准化后可以筛选出更多的差异甲基化基因。值得强调的是,在一套数据集中,经过分位数标准化后额外找到的差异甲基化基因,与在同种癌型的独立数据集中的原始信号的改变方向有高度的一致性,具有显著的非随机性,反映了这些差异甲基化基因在独立数据中存在着明确的生物信号。这些结果说明,采用分位数标准化方法,可以提高识别差异甲基化基因的统计效能。因此,在正常样本和癌症样本中,对于信号值的中值无显著差别的甲基化谱数据,建议采用针对基因表达谱数据处理开发的标准化方法。但是,在结肠癌、肺癌和肾癌中,发现有1%经过分位数标准化后筛选出的差异甲基化基因,其甲基化状态与原始数据中信号的改变方向相反。因此,数据标准化过程也可能会产生过标准化现象,从而导致产生1%左右的假阳性结果,需要将其去除。
分析也显示,许多基因仅在研究同种癌型的一套数据中发生了显著的甲基化改变,而未能够在另外一套数据集中被识别为差异甲基化。这种表面的低重复性,可能来源于甲基化变化在个体间的生物学变异与异质性、严格的统计控制与较小的样本量所产生的低统计效能等因素[21,27]。特别地,在癌基因组中,异常的DNA甲基化改变可以被当作生物标记,用来预测癌症后果和寻找药物靶点[3];而统计效能的下降,会导致在不同数据中选择少数最显著的基因作为标记变得很不稳定[29]。为了评价来自研究同种癌型的不同实验所识别的最显著的差异甲基化基因的可重复性,有必要考虑它们的功能相关关系,以及甲基化改变方向的一致性,而不是简单地计算它们本身的重合率[30]。值得注意的是,实验设计的缺陷如实验中存在批次效应(即与生物学无关因素的变异和样本类别显著相关),很可能会导致不可靠的生物学结论[4]。例如,在错误地整合存在批次效应的不同批次的甲基化数据时,很可能会错误地判断差异甲基化基因的改变方向[4]。因此,应该更加注重优化实验设计,严格地使技术变异随机分布在不同的生物组中,并且在各组中检测足够多的样本[31]。
[1]Melnikov A, Scholtens D, Godwin A, et al. Differential methylation profile of ovarian cancer in tissues and plasma[J].J Mol Diagn,2009,11(1):60-65.
[2]Shaknovich R,Geng H,Johnson NA,et al.DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma[J].Blood,2010,116(20):e81-e89.
[3]Lugthart S,Figueroa ME,Bindels E,et al.Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1[J].Blood,2011,117(1):234-241.
[4]Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data[J].Nat Rev Genet,2010,11(10):733-739.
[5]Quackenbush J. Microarray data normalization and transformation[J].Nat Genet,2002,32 Suppl:496-501.
[6]Larsson O,Wennmalm K,Sandberg R.Comparative microarray analysis[J].OMICS,2006,10(3):381-397.
[7]Bolstad BM,Irizarry RA,Astrand M,et al.A comparison of normalization methods for high density oligonucleotide array data based on variance and bias[J].Bioinformatics,2003,19(2):185-193.
[8]Ron-Bigger S,Bar-Nur O,Isaac S,et al.Aberrant epigenetic silencing of tumor suppressor genes is reversed by direct reprogramming[J].Stem Cells,2010,28(8):1349-1354.
[9]Wang Dong,Cheng Lixin,Wang Mingyue,et al.Extensive increase of microarray signals in cancers calls for novel normalization [J]. Computational Biology and Chemistry,2011,35(3):126-130.
[10]Calza S, Valentini D, Pawitan Y. Normalization of oligonucleotide arrays based on the least-variant set of genes[J].BMC Bioinformatics,2008,9:140.
[11]Esteller M.Epigenetics in cancer[J].N Engl J Med,2008,358(11):1148-1159.
[12]Feinberg AP,Vogelstein B.Hypomethylation distinguishes genes of some human cancers from their normal counterparts[J].Nature,1983,301(5895):89-92.
[13]Christensen BC,Houseman EA,Marsit CJ,et al.Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context[J].PLoS Genet,2009,5(8):e1000602.
[14]O'Riain C,O'shea DM,Yang Y,et al.Array-based DNA methylation profiling in follicular lymphoma[J].Leukemia,2009,23(10):1858-1866.
[15]Rakyan VK, Down TA, Maslau S, et al. Human agingassociated DNA hypermethylation occurs preferentially at bivalent chromatin domains[J].Genome Res,2010,20(4):434-439.
[16]Teschendorff AE,Menon U,Gentry-Maharaj A,et al.Agedependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer[J].Genome Res,2010,20(4):440-446.
[17]Barrett T,Troup DB,Wilhite SE,et al.NCBI GEO:archive for high-throughput functional genomic data[J].Nucleic Acids Res,2009,37(Database Issue):D885-D890.
[18]Comprehensive genomic characterization defines human glioblastoma genes and core pathways[J].Nature,2008,455(7216):1061-1068.
[19]Reimers M.Making informed choices about microarray data analysis[J]. PLoS Computational Biology,2010,6(5):e1000786.
[20]Benjamini Y,Hochberg Y.Controlling the false discovery rate:a practical and powerful approach to multiple testing[J].Journal of the Royal Statistical Society. Series B(Methodological),1995,57(1):289-300.
[21]Zhang Min,Yao Chen, Guo Zheng,et al.Apparently low reproducibility of true differential expression discoveries in microarray studies[J].Bioinformatics,2008,24(18):2057-2063.
[22]Rocke DM, Ideker T, Troyanskaya O, et al. Papers on normalization,variable selection,classification or clustering of microarray data[J].Bioinformatics,2009,25(6):701.
[23]Ivanova AV,Ivanov SV,Prudkin L,et al.Mechanisms of FUS1/TUSC2 deficiency in mesothelioma and its tumorigenic transcriptional effects[J].Mol Cancer,2009,8:91.
[24]Guervos MA,Marcos CA,Hermsen M,et al.Deletions of N33,STK11 and TP53 are involved in the development of lymph node metastasis in larynx and pharynx carcinomas[J].Cell Oncol,2007,29(4):327-334.
[25]Kawakami K, Hirata H, Yamamura S, et al. Functional significance of Wnt inhibitory factor-1 gene in kidney cancer[J].Cancer Res,2009,69(22):8603-8610.
[26]Urakami S,Shiina H,Enokida H,et al.Wnt antagonist family genes as biomarkers for diagnosis,staging,and prognosis of renal cell carcinoma using tumor and serum DNA[J].Clin Cancer Res,2006,12(23):6989-6997.
[27]Carvajal-Rodriguez A,de Una-Alvarez J,Rolan-Alvarez E.A new multitest correction(SGoF)that increases its statistical power when increasing the number of tests[J]. BMC Bioinformatics,2009,10:209.
[28]Laird PW.Principles and challenges of genome-wide DNA methylation analysis[J].Nat Rev Genet,2010,11(3):191-203.
[29]Deng Dajun,Liu Zhaojun,Du Yantao.Epigenetic alterations as cancer diagnostic,prognostic,and predictive biomarkers[J].Adv Genet,2010,71:125-176.
[30]Zhang Min, Zhang Lin, Zou Jinfeng, et al. Evaluating reproducibility of differential expression discoveries in microarray studies by considering correlated molecular changes[J].Bioinformatics,2009,25(13):1662-1668.
[31]Ein-Dor L,Zuk O,Domany E.Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer[J].Proc Natl Acad Sci U S A,2006,103(15):5923-5928.
