农杆菌介导法构建里氏木霉ura3基因突变体的研究
2011-08-08闫作梅
闫作梅,张 旭,李 杰
(东北农业大学生命科学学院,哈尔滨 150030)
里氏木霉作为工业菌株,常用来生产多种重要的酶类,包括纤维素酶、半纤维素酶、蛋白酶、淀粉酶等,具有巨大的商业价值[1-2]。目前,里氏木霉的基因组测序已经完成,对于其基因的功能有待研究和开发[3]。同时,里氏木霉是理想的表达真核基因的系统,具有很好的合成和分泌蛋白的能力,在理想的培养条件下发酵分泌纤维素酶可达到40~100 g·L-1发酵液[4-5],被称为重组蛋白工厂。Harkki等报道,通过基因定位置换整合使编码里氏木霉CBHI的基因失活,结果EGI的产量提高,不再产生CBHI;1993年,Karhunen等将编码里氏木酶EGI的基因置于强启动子CBHI的控制下,用此表达盒替换染色体的CBHI位点之后,EGI产量是通常强纤维素分解菌所产CBHI量的2倍或者与其一样多;此外,还有一些关于里氏木酶的一个或几个纤维素酶基因经基因定位置换整合而失活的报道[6-7]。
在里氏木霉转化研究中,常以营养缺陷型互补基因作为筛选标记,这种筛选标记易于筛选获得转化子,被广泛应用到丝状真菌的转化系统中[8]。其中乳清酸核苷-5-磷酸脱羧酶(ura3)基因缺陷株及其相应的转化系统己被证明是一种行之有效的转化系统。由于5-氟乳清酸(5-FOA)对原养型菌株有毒性作用,而ura3缺陷菌株则对其产生抗性,所以提供了一种可以正反双向筛选的筛选系统。
本研究尝试利用农杆菌介导法通过同源重组获得里氏木霉ura3基因缺失突变体,从而为里氏木霉转化系统研究提供材料。
1 材料与方法
1.1 材料
1.1.1 菌株和培养条件
大肠杆菌培养用LB培养基,37℃培养;里氏木霉菌种活化用土豆培养基,培养里氏木霉菌丝体用PDA培养基或MM培养基,28℃培养。
1.1.2 菌株和质粒
菌株:大肠杆菌DH5α、农杆菌GV3101、里氏木霉QM9414。
质粒:pEMT-5由本实验室保存。1.1.3 分子试剂
胶回收试剂盒Gel Extract Kit(购自百泰克公司)。限制性核酸内切酶、T4DNA连接酶、rTaq酶、LA-Taq酶 、 dNTP、 PyrobestTMDNA Polymerase、pMD18-T vector等(购自大连宝(TaKaRa)生物工程有限公司)。
1.1.4 PCR引物设计
ura3引物序列如下:

pyrA引物序列如下:

ura3和pyrA基因农杆菌PCR检测引物序列:


1.2 方法
1.2.1 ura3和pyrA基因的克隆
以里氏木霉QM9414基因组DNA为模板,PCR扩增ura3基因片段,预期扩增片段大小为1 469 bp。PCR反应条件:94℃预变性10 min,94℃变性1 min,55℃退火30 s,72℃延伸1 min,72℃延伸10 min,4℃保存,30个循环。
以黑曲霉UV48基因组DNA为模板,PCR扩增pyrA基因片段,大小为3 370 bp。PCR反应条件:94℃预变性10 min,94℃变性1 min,55℃退火30 s,72℃延伸4 min,72℃延伸10 min,4℃保存,30个循环。
1.2.2 ura3缺失基因载体和pyrA基因载体构建
质粒提取、酶切、转化参见文献[9],连接参见文献[10],DNA片段回收参见胶回收试剂盒说明。
1.2.3 重组质粒转化根癌农杆菌
取0.5~1 μL重组质粒DNA,加入100 μL根癌农杆菌感受态细胞中,轻轻混匀;冰浴5 min,液氮冷冻8 min,迅速至37℃温浴融化;加入800 μL YEB液体培养基,28℃,轻轻摇动4~5 h;
5 000 r·min-1离心5 min,倒掉大部分上清,将剩余约50 μL菌液用移液器转至含有Km 100 mg·L-1、Gent 50 mg·L-1、Rif 50 mg·L-1的 YEB 固体培养基上涂匀。28℃暗培养48~96 h。
1.2.4 根癌农杆菌和里氏木霉共培养
用5 mL灭菌蒸馏水从培养5~7 d的PDA平板上洗下里氏木霉菌的分生孢子,用MM液体培养基稀释至相应的孢子浓度。接种含双元载体系统的pEMT-△ura3根瘤农杆菌单菌落于3 mL液体YEB培养基(含 50 mg·L-1Rif、50 mg·L-1Gent、100 mg·L-1Kan)中,200 r·min-1,28 ℃培养 2~3 d,离心收集菌体,用一定体积的IM液体培养基重悬菌体。将菌液浓度调到660 nm处的吸收值为0.25,继续培养6 h,OD660达到0.6~0.8。取处理好的里氏木霉孢子悬液和根瘤农杆菌菌液各100 μL,混匀,涂布MM 培养基(含 200 μmol·L-1乙酰丁香酮),24 ℃共培养48 h。
1.2.5 转化子的筛选
将在24℃孵育48 h的培养物移至含有尿嘧啶核苷(1.87 mg·mL-1)、五氟乳氰酸(5 mg·mL-1)和阿莫西林(100 mg·mL-1)的诱导培养基上28℃培养8~10 d至抗性菌落出现。
1.2.6 转化子的鉴定
提取转化子的基因组DNA,PCR鉴定转化子。如果ura3基因被敲除,扩增出片段应为445 bp;如果黑曲霉的pyrA插入到里氏木霉的基因组中,扩增出650 bp的片段。
2 结果与分析
2.1 5-FOA筛选浓度的确定
将里氏木霉孢子接种于含不同浓度5-FOA的PDA固体培养基上,28℃培养7 d后,里氏木霉生长情况见图1。5-FOA浓度在5 mg·mL-1时即可有效抑制里氏木霉的生长,因此确定5-FOA筛选浓度为 5 mg·mL-1。
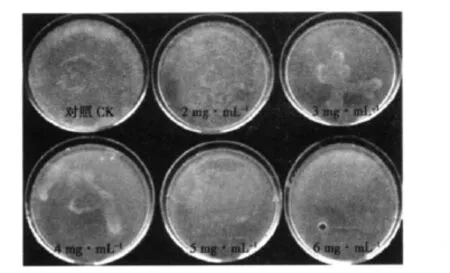
图1 5-FOA对里氏木霉生长的抑制作用Fig.1 Inhibition effect of 5-FOA on the germination and growth of Trichoderma reesei strains
2.2 ura3和pyrA基因的克隆
ura3基因PCR扩增得到一条1 469 bp的目的条带(见图2);pyrA基因PCR扩增得到一条3 300 bp的目的条带(见图3)。将上述扩增条带克隆至T载体,筛选阳性克隆送交测序。测序结果表明,已获得完整的ura3基因和pyrA基因片段(序列略)。
2.3 ura3缺陷型载体的构建
ura3缺陷型载体pEMT-△ura3的构建过程见图4。HindⅢ、XbaⅠ双酶切质粒pMD-ura3,回收约2 600和1 470 bp的片段。再将1 470 bp的小片段用SmaⅠ酶切,回收约650和700 bp片段。将上述3个片段连接,获得质粒pMD-△ura3。经酶切和测序证明,该载体上的ura3基因缺失110 bp,结果正确。再用Eco RⅠ、NheⅠ双酶切质粒pEMT-5,回收约8 000 bp的载体片段,与经同样酶切质粒pMD-△ura3回收的1 350 bp的目的片段连接,获得载体pEMT-△ura3。通过冻融法将pEMT-△ura3转入农杆菌GV3101,转化菌落的PCR鉴定结果见图5。

图2 ura3基因PCR扩增结果Fig.2 Results of PCR amplification of ura3

图3 pyrA基因PCR扩增结果Fig.3 Results of PCR amplification of pyrA
2.4 黑曲霉pyrA基因载体的构建
黑曲霉pyrA基因载体pEMT-pyrA的构建过程见图6,将pEMT-ku70载体用XbaⅠ、Sna BⅠ双酶切,回收11 524 bp的载体片段,然后用XbaⅠ,NcoⅠ双酶切的黑曲霉pyrA基因片段与载体连接,构建重组质粒pEMT-pyrA。

图4 pEMT-△ura3载体的构建Fig.4 Construction of vector pEMT-△ura3

图5 pEMT-△ura3转入农杆菌的鉴定结果Fig.5 Characterization of pEMT-△ura3 transfer in Agrobacterium

图6 pEMT-pyrA载体的构建Fig.6 Construction of vector pEMT-pyrA
通过冻融法将pEMT-△ura3转入农杆菌GV3101,转化菌落的PCR鉴定结果见图7。
2.5 ura3基因缺失突变株的分子鉴定
通过农杆菌介导法将pEMT-△ura3转化里氏木霉QM9414,将筛选平板上获得的菌落进行PCR验证(见图 8)。
结果显示随机挑选的20个转化子中只有1个可以扩增得到ura3的缺失基因(445 bp,泳道13),即发生的是同源重组,将其命名为QM9414-△ura3突变株。还有5株(泳道3、7、9、10、12)同时扩增出445和555 bp特异条带,即发生的是非同源重组,其同源重组率为16.6%(1/6)。

图7 pEMT-pyrA转入农杆菌的鉴定结果Fig.7 Characterization of pEMT-pyrA transfer in Agrobacterium
2.6 里氏木霉ura3基因缺失突变体的回复
采用农杆菌介导法将pyrA基因转化里氏木霉QM9414-△ura3突变株,在不含有尿嘧啶核苷酸的基本培养基平板上筛选回复突变株。然后应用PCR筛选整合有pyrA基因的转化子,在随机挑取的30个转化子中,有3个扩增出目的基因(见图9)。

图8 △ura3基因缺失突变株的PCR检测Fig.8 PCR detection of△ura3 gene disruptant

图9 pyrA转化子的PCR检测Fig.9 PCR detection of pyrA gene
3 讨论
目前,常用紫外诱变的方法对丝状真菌的菌株进行改造,但是不易控制诱变的位点。而同源重组利用DNA转化技术,通过载体DNA序列与靶细胞内染色体上同源DNA序列间的重组从而定向改变细胞遗传特性,具有明显的优势[8]。
由于丝状真菌遗传转化系统和选择标记被成功的应用于许多的真菌转化中,这就为人们在基因水平上研究真菌的遗传背景和菌株的改造提供了有利的条件,其中以根癌农杆菌介导的遗传转化体系备受关注[11-12]。根癌农杆菌介导的丝状真菌转化与传统的转化方法相比具有以下优点[13-16]:①转化效率高;②可导入大片段的DNA;③导入基因拷贝数低,表达效果好,稳定遗传;④农杆菌转化方法使用的技术、仪器简单;⑤同源重组效率高。Lima等利用传统的PEG介导的方法对T.atroviride进行转化,同源重组的频率很低,而利用农杆菌介导的方法获得了60%的同源重组的转化子,使得其敲除基因的效率可达14%~75%,成功实现了对tmk1和tga3基因非编码区的基因破坏[17]。
本试验全面考虑了高效基因修饰和置换技术的要点,有机整合了一些相关技术,通过农杆菌介导法提高里氏木霉的转化效率,通过以ura3作为筛选标记降低了转化子的假阳性,提高导入DNA片段的同源重组效率。所建立的高效基因修饰和置换系统符合食品级表达系统的要求,由此所研制的工程菌和重组蛋白具有高度的安全性,可用于食品加工,应用领域更为广阔。
本研究利用农杆菌介导方法将带有△ura3基因的载体导入到里氏木霉宿主细胞内,使载体上的△ura3基因与宿主细胞的染色体DNA发生同源重组,从而获得以ura3作为筛选标记的营养缺陷菌株,同时证实农杆菌介导的方法能高效的敲除里氏木霉基因,为研究里氏木霉的功能基因提供了高效的、快速的方法。
4 结论
本研究从里氏木霉QM9414中克隆了ura3基因,构建了ura3基因敲除载体pEMT-△ura3。从黑曲霉中克隆了pyrA基因,构建了pyrA基因载体pEMT-pyrA。采用农杆菌介导法将ura3缺失基因转化里氏木霉QM9414,筛选获得QM9414-△ura3突变株,其同源重组率为16.6%。将pyrA基因转化QM9414-△ura3突变株,筛选获得回复突变株。
[1] Nevalainena K M H,Teoa V S J.Enzyme production in industrial fungi:molecular genetic strategies for integrated strain improvement[J].Applied Mycology and Biotechnology,2003(3):241-259.
[2] Punt P J,van Biezen N,Conesa A,et al.Filamentous fungi as cell factories for heterologous protein production[J].Trends Biotechnol,2002,20(5):19-33.
[3] 李锐,贺亮,邓旭明.丝状真菌蛋白质组分析[J].东北农业大学学报,2008,39(5):58-61.
[4] Levin A M,de Vries R P,W sten H A B.Localization of protein secretion in fungal coloies using a novel culturing technique;thering-plate system[J].Journal of Microbiogical Methods,2007,69(2):399-401.
[5] Wang T H,Wu J,Zong Y X.Advances of molecular biology of Trichoderma reesei[J].Mycosystema,2000,19(1):147-152.
[6] Nevalainen K M H,Valentino S J T,Bergquist P L.Heterologous protein expression in filamentous fungi[J].Trends Biotechnol,2005,23(9):468.
[7] Nayak T,Szewczyk E,Oaklay C E,et al.A versatile and efficient gene targeting system for Aspergillus nidulans[J].Genetics,2005(12):30.
[8] Degefu Y,Hanif M.Agrobacterium tumefaciens-mediated transformation of Helminthosporium turcicum,the maize leaf blight fungus[J].Arch Microbiol,2003,108:279-284.
[9] 萨姆布鲁克等.分子克隆实验指南[M].2版.北京:科学出版社,1992.
[10] 李杰,李晶,朱延明,等.DREB1A基因植物表达载体的构建[J].东北农业大学学报,2003,34(2):199-204.
[11] Ninomiya Y,Suzuki K,Ishii C,et al.Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining[J].Proc Natl Acad Sci,2004,101:12248-12253.
[12] Ruiz-Diez B.Strategies for the transformation of filamentous fungi[J].J APPl Microbiol,2002,92:189-195.
[13] Gouka R J,Gerk C,Hooykaas P J,et al.Transformation of Aspergillus awamori by Agrobacterium tumefaciens-mediated homologous recombination[J].Nat Biotechnol,1999,17:598-601.
[14] Fang W,Pei Y,Bidochka M J.Transformation of Metarhizium anisopliae mediated by Agrobacterium tumefaciens[J].Can J Microbiol,2006,52:623-626.
[15] Godio R P,Fouces R,Gudina E J,et al.Agrobacterium tumefaciens-mediatedtransformationoftheantitumorclavaricacidproducing basidiomycete Hypholoma sublateritium[J].Curr Genet,2004,46:287-294.
[16] 王敬国,张卫国,夏林,等.黑曲霉原生质体转化效率影响因子研究[J].东北农业大学学报,2007,38(4):20-25.
[17] Lima I G,Duarte R T,Furlaneto L,et al.Transformation of the entomopathogenic fungus Paecilomyces fumosoroseus with Agrobacterium tumefaciens[J].Lett Appl Microbiol,2006,42:631-636.
