一氧化氮在脑缺血预处理中的作用及其机制
2011-08-06张珍珍
张 超, 季 晖, 赵 倩, 张珍珍
(中国药科大学药理学教研室,江苏 南京 210009)
脑缺血预处理(ischemic preconditioning,IPC)是指脑组织在经过非致死性短暂的重复缺血后,能增强其对缺血的耐受性的一种现象[1]。短暂性脑缺血诱导的脑IPC效应是一种多因素级联反应,可促进脑内神经元存活,减轻缺血后损伤。一氧化氮(NO)可调节大脑血流量和局部大脑灌注时的新陈代谢活性,对IPC的产生起着至关重要的作用。
1 一氧化氮与一氧化氮合酶
1.1 在脑内的功能
NO由一氧化氮合酶(NOS)产生,NOS具有3种亚型:神经型(nNOS)、内皮型(eNOS)和诱导型(iNOS)。在健康的大脑中,NO主要由神经元内的nNOS和内皮上的eNOS产生。在神经元中,nNOS主要存在于细胞质中,激活后迁移至细胞膜上,与谷氨酸盐依赖的N-甲基-D-天冬氨酸(NMDA)受体结合并激活受体[2]。NMDA受体被激活后,引起Ca2+内流,激活nNOS产生较低水平的NO[3]。在损伤、缺氧等应激条件刺激下,可诱导胶质细胞产生iNOS,且不依赖Ca2+而持续产生并释放NO[4-5]。有研究者认为在线粒体中存在着一种新的NOS——线粒体NOS(mtNOS),但人们对此存在争议,有人认为所谓mtNOS可能只是线粒体中的nNOS[6]。
NO可介导谷氨酸盐信号,并对细胞-细胞间的信号传导产生影响,而谷氨酸盐信号在长期增强或者减弱的过程中,NO可能发挥一个神经兴奋传递的反馈传感器作用,这种反馈对于突触前/后膜的兴奋传递和神经网的活性很重要。作为内皮细胞衍生舒张因子,由eNOS和nNOS产生的NO可通过调节局部大脑血流量增加神经元活性。
1.2 在脑缺血后的作用
研究表明,健康脑内的NO浓度是纳摩尔水平,然而脑缺血后其浓度升至微摩尔水平,这缘于nNOS的激活。NOS在脑缺血后的作用可被分为两个方面,即调节血管功能(由eNOS介导)和产生毒性作用(初期由nNOS介导,后期由iNOS介导)。
有研究者曾在nNOS基因敲除小鼠实验中发现,nNOS基因敲除小鼠在脑缺血后脑内NO水平没有增加,并对局部缺血和全脑缺血产生耐受。脑缺血诱导nNOS合成的NO对组织的损伤机制包括:致聚ADP-核糖聚合酶(PARP)激活,导致细胞储存能量耗尽[7];与超氧化物结合生成亚硝酸盐;诱导细胞凋亡(Elibol等,Neuroscience,2001 年)。与nNOS基因敲除小鼠实验的结果一致,nNOS抑制剂也可使动物模型对缺血产生耐受(Yoshida等,J Cereb Blood Flow Metab,1994 年)。
已有研究表明,在脑缺血开始阶段,iNOS在脑内并无表达,直到几天后,胶质细胞和炎症细胞进入了缺血核心区,方才有表达,而由iNOS产生的NO则可加重组织损伤。Ono等[8]证实小胶质细胞中因iNOS激活所产生并释放的NO与缺血后延迟性神经元损伤密切相关,且使用iNOS特异性抑制剂或在iNOS基因敲除模型小鼠中进行的实验也证明,抑制iNOS的表达,可减轻这种损伤(Iadecola等,Neuroscience,1997 年)。
与nNOS和iNOS的毒性作用不同,eNOS在脑缺血后可发挥脑血管保护作用,即舒张血管和抑制血小板聚集及白细胞与内皮细胞黏附。实验表明,在脑缺血后,eNOS基因敲除小鼠与野生型小鼠相比,其脑内有更大的核心缺血区和更小的半影区,这可能缘于其缺失eNOS的脑血流量调节作用(Huang等,J Cereb Blood Flow Metab,1996年)。
2 脑缺血预处理机制
IPC可出现在不同的组织中,如心脏、大脑、肝、肺和胃肠道片段[9-10],但是其在各组织中的激活和调控机制是否一样尚不得而知。IPC的一些保护机制已被证实,如改变细胞基因、上调热休克蛋白、减少脂过氧化反应、促进炎症和线粒体新陈代谢等[11-15],但其分子机制也不十分清楚。
依据短暂性缺血后产生保护效应的时间间隔的不同,脑IPC可分为两种类型:即时型(early preconditioning)和延迟型(late preconditioning),即 IPC的保护效应呈“双峰”时相。前者在短暂性缺血后数分钟至3 h内即产生保护效应,而后者的保护效应则出现在短暂性缺血后24~72 h,两者的病理生理及保护机制不同。即时型IPC因为在短暂性缺血后很快产生,仅被认为是翻译前水平的调控,不像延迟型IPC涉及到基因表达的改变,因此其保护效应产生的时间较短。
对脑IPC的研究起初采用了沙土鼠的全脑缺血模型,结果显示,阻断双侧颈总动脉达5 min,即可造成海马CA1区的神经元损伤,而若在此前预先致脑形成2 min的短暂性缺血,则可减轻随后缺血造成的CA1区神经元损伤,提示短暂性缺血可对脑组织产生保护效应;但这种保护效应在1~3 d后才出现,说明这是一种延迟型IPC效应。最新研究表明,NMDA受体的激活和相关蛋白的合成是延迟型IPC产生的必要条件[16]。
尽管IPC确切的信号分子尚不明确,但其延迟保护效应的一些激活分子已被人们发现,包括腺苷、缓激肽、细胞因子、NO和活性氧(ROS)。这些分子可以激活IPC的上游信号,其中有些分子与NOS的激活相关,提示NO的产生可能是延迟型IPC效应产生的关键。
3 对一氧化氮在脑缺血预处理中作用的研究
3.1 体外模型实验研究
缺氧-缺糖(OGD)模型可用于对原代神经元或PC12细胞进行的体外模拟脑缺血研究。在神经元细胞培养模型中,短暂的OGD可诱导IPC效应,表现为神经元的抗氧化应激能力增强。然而,细胞培养模型只能用来研究神经元产生IPC的内源性通路[17],并不能阐明血管的作用和不同组织结构之间的相互作用对IPC产生的影响。
体外实验显示,神经元细胞经5~30 min的OGD处理后,对24 h后45~55 min的OGD损伤有保护作用(Grabb等,J Neurosci,1999年)。这种IPC效应与NMDA受体激活、Ca2+内流和新蛋白合成相关,而L-NAME可阻断此IPC效应,提示NO参与了此IPC效应的产生。使用原代皮层细胞培养模型进行的实验还发现,NMDA受体兴奋后,激活nNOS,导致NO水平升高,继而激活可抑制Ras相关信号传导的p21 Ras,产生IPC效应,而使用p21 Ras显性失活突变体或者p21 Ras抑制剂可以阻断IPC效应,提示下调Ras相关信号,包括Raf/MEK/ERK通路传导,可能改变基因表达,并刺激神经元生长和存活相关程序,从而诱导IPC效应(Gonzalez-Zulueta等,Proc Natl Acad Sci USA,2000 年)。
3.2 体内模型实验研究
小鼠实验显示,连续将大脑中动脉阻断(MACO)3次(每次持续5 min),对30 min后永久性脑缺血损伤有保护作用,24 h后的脑梗死体积检测验证了这一点(Stagliano等,J Cereb Blood Flow Metab,1999 年)。Atochin等[18]在 eNOS 和 nNOS 基因敲除及野生型小鼠实验中采用同样的方法考察了eNOS和nNOS在IPC中的必要性,其间采用多普勒血流仪记录血流量变化。结果表明,野生型小鼠经过3次MACO后,可使永久性脑缺血24 h后的脑梗死体积减少,但eNOS和nNOS基因敲除小鼠却未见有此IPC效应。
在新生小鼠缺氧-缺血模型中,IPC效应可以被非特异性NOS抑制剂L-精氨酸(L-NAME)而并非nNOS特异性抑制剂7-硝基吲哚(7-nitroindazole)和iNOS特异性抑制剂氨基胍(aminoguanidine)所拮抗(Gidday 等,J Cereb Blood Flow Metab,1999 年)。Gustavsson等[19]在同一模型试验中也证实eNOS的基因表达与脑IPC效应相关,即eNOS基因表达的增加可致大脑血管舒张及血流量增加,从而产生IPC效应。
大鼠实验显示,给予3 min短暂的MACO或脂多糖(LPS),可产生脑IPC效应。其中,由LPS诱导的IPC效应被认为与eNOS表达的增加有关,可被非特异性NOS抑制剂L-NAME阻断,然而,L-NAME却不能抑制短暂性MACO所致IPC效应(Puisieux等,Eur J Pharmacol,2000年)。这一实验结果与之前人们的研究结果出现偏差,究其原因,可能是实验动物种群差异(大鼠 vs小鼠)、模型差异(3 min MACO 1次 vs 5min MACO 3次)和L-NAME不能有效阻断eNOS表达所致。eNOS诱导脑IPC效应的可能机制是,舒张大脑血管,增加缺血半影区血流量,从而减小脑梗死体积;且与干预白细胞-内皮细胞以及血小板-内皮细胞间相互作用有关(Jiang等,Brain Res,2002 年)。
挥发性麻醉剂也可诱导脑 IPC效应,Wang等[20]认为这和蛋白激酶B(Akt)的激活以及三磷酸腺苷(ATP)依赖性通道等与NO密切相关的信号通路有关。给大鼠腹腔注射异氟烷或者氟烷后24 h,再行2 h MACO,则可产生脑IPC效应,而iNOS抑制剂氨基胍可阻断此IPC效应,提示iNOS参与了这些麻醉剂诱导IPC效应产生的过程(Kapinya等,Brain Res,2000 年)。Kitano 等[21]在小鼠实验中发现,使用异氟醚诱导脑IPC效应,存在性别差异,即此诱导方法只对雄性小鼠有效,而对于雌性小鼠无效。这种性别差异来自于Akt的活性差异,继而改变下游eNOS的活性。
综上所述,NO在IPC中的作用可分为以下两个方面:第一,可能通过内源性通路参与IPC效应,发挥触发器和调控器双重作用,调节神经元对脑缺血的耐受;第二,作为内皮细胞衍生舒张因子,可在脑缺血后增加灌注血流量,抑制内皮细胞、血小板和白细胞的相互黏连,从而降低脑缺血所致血管功能性损伤。
4 对一氧化氮在脑缺血预处理中作用机制的研究
已有研究表明NO在IPC中的作用机制涉及以下途径:1)Ras/Raf/MEK/ERK和PI3K/Akt通路,NO可激活这两条通路,并产生级联反应,如细胞骨架蛋白磷酸化、细胞黏附分子磷酸化以及转录因子[如Elk-1、cAMP反应元件结合蛋白(CREB)、NF-κB等]的激活,而转录因子的激活又可以导致基因表达的变化,触发 IPC效应;2)NF-κB通路,NF-κB参与了由细胞因子和生长因子引起的细胞分化、凋亡和生存过程,而短暂性脑缺血诱导产生的NO可激活NF-κB,并诱导产生iNOS,引发IPC效应;3)线粒体K+-ATP通道,一种ATP敏感性的K+通道,被认为可能是延迟型IPC的效应器,而NO可以选择性激动线粒体K+-ATP通道。
4.1 Ras/Raf/MEK/ERK通路
Ras/Raf/MEK/ERK通路可调节细胞增殖、分化、存活、凋亡的基本过程。研究表明,由nNOS产生的NO可激活G蛋白Ras(Gonzalez-Zulueta等,Proc Natl Acad Sci USA,2000 年),使之结合 Ras激酶,并迁移到细胞膜表面;Ras激活丝苏氨酸激酶Raf,继而激活MEK激酶家族(MAPK/ERK激酶,MEK1和MEK2),MEK又致ERK激酶家族(ERK1和ERK2,即p42和p44)磷酸化并激活,而ERK激酶是这个级联反应的效应分子,不仅可致细胞质中靶蛋白磷酸化,还可致核蛋白和转录因子,如CREB和Elk-1磷酸化(见图1)[16]。此外,体外神经元培养模型实验发现,ERK的激活与 IPC效应相关(Kolch等,Biochem J,2000年)。笔者所在课题组进行的大鼠实验也显示,IPC模型大鼠在脑缺血再灌注后脑内磷酸化ERK(p-ERK)水平升高,而其接受新型NO供体型化合物(S)-ZJM-289治疗后,IPC效应增强,且其脑内p-ERK表达水平进一步提高,提示(S)-ZJM-289增强IPC效应的作用可能与ERK蛋白的激活相关。
4.2 PI3K/Akt通路
NO激活的Ras也可通过促进生长因子与酪氨酸激酶受体或者G蛋白偶联受体结合而激活PI3K/Akt通路[15]。3-磷酸磷脂酰肌醇激酶(PI3K)被激活后,可水解细胞膜上 4,5-二磷酸磷脂酰肌醇(PIP2)生成第2信使 PIP3,而PIP3与细胞内含有PH结构域的信号蛋白Akt和磷酸肌醇依赖性蛋白激酶(PDK)结合,促使PDK致Akt磷酸化并激活(见图1)。
Akt也称蛋白激酶B(PKB),是一种相对分子质量为60000的丝氨酸/苏氨酸激酶,具有3种亚型,其中 Akt1是与大脑功能密切相关的一种亚型[22],可在 4 个位点(Ser124、Thr308、Thr450 和Ser473)被PDK磷酸化,且Thr308和Ser473位点的磷酸化可增强Akt1激酶活性。Akt1激活后可致丝氨酸和苏氨酸磷酸化,继而影响诸多靶点,如基因转录调控蛋白(如 FOCO、IKK、mdm2等)、细胞质酶、组蛋白和细胞程序化死亡相关蛋白(如BAD、Bax、Caspase 9等)。
细胞内钙浓度的增加可以激活nNOS和eNOS。最近研究表明,eNOS的活性也受磷酸化的苏氨酸和丝氨酸残基调控,如因Ser1179位点磷酸化而激活以及因Thr479位点磷酸化而失活[23]。Akt激酶可致eNOS在Ser1179位点磷酸化并激活,是重要的体内eNOS调控因子,其介导的eNOS磷酸化改变了细胞对生长因子如胰岛素样生长因子(IGF-1)的应答。除Akt以外,其他激酶,如PKA、AMPK和PKC也可致 eNOS在不同位点磷酸化,调控其活性。eNOS活性的增加对于IPC的影响机制包括:调节Caspase的表达和磷酸化、PKC通路的激活及基因表达的改变。
总的来说,Akt1及其下游信号分子可促进脑缺血再灌注后神经元的存活,与IPC效应的产生密切相关。
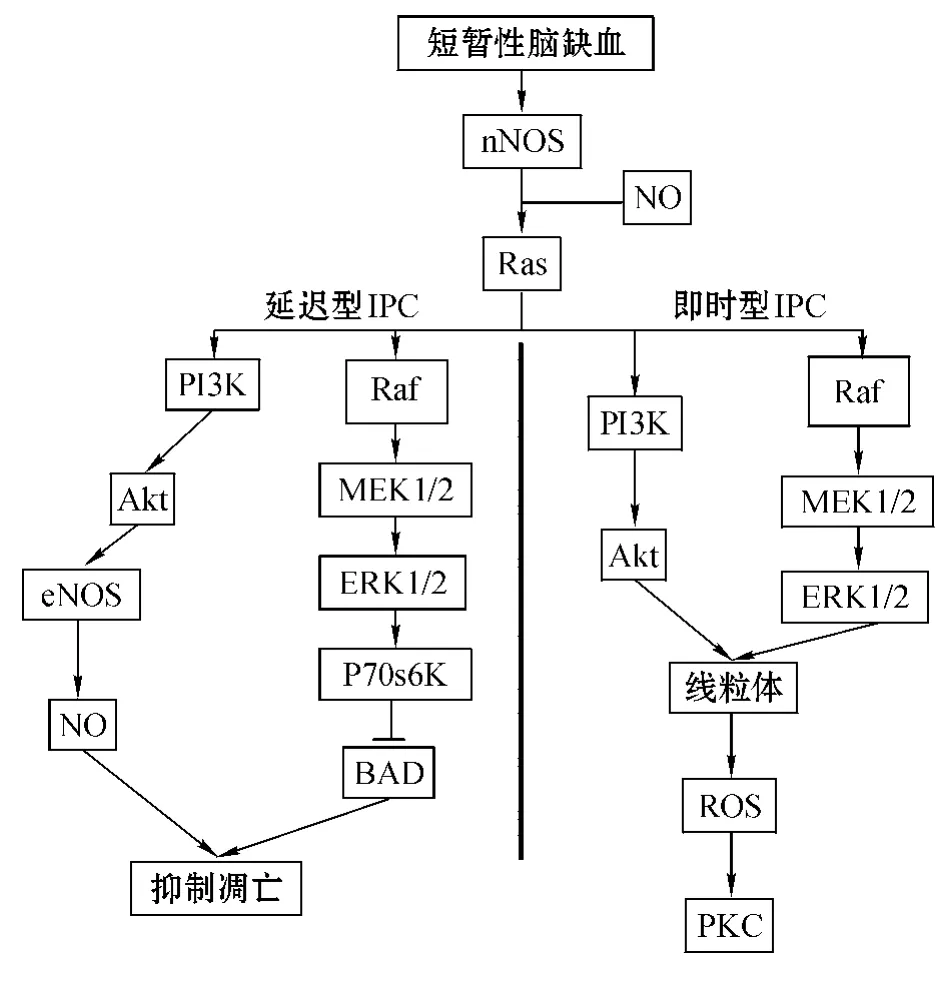
图1 NO经Ras/Raf/MEK/ERK和PI3K/Akt通路诱导IPC的分子机制Figure 1 Molecular mechanism of NO-induced IPC via Ras/Raf/MEK/ERK and PI3K/Akt signal pathways
4.3 NF-κB 通路
在正常状态下,NF-κB在胞质中与其抑制因子I-κB结合而失去转录活性。短暂性脑缺血后产生的适量的NO则能促进PKC致IKK(I-κB的激酶)磷酸化并激活,导致 I-κB磷酸化及降解,使其与NF-κB 解离[24-25],被释放的 NF-κB 转移到细胞核内并诱导目的基因表达 iNOS,激活 CREB[16],并产生IPC效应(见图2)。笔者所在课题组研究发现,(S)-ZJM-289用于IPC模型大鼠时,可改善大鼠的神经行为学功能和脑损伤指标。且免疫组化和Western Blot实验显示,给IPC模型大鼠使用了(S)-ZJM-289后,大鼠脑内NF-κB和eNOS蛋白表达上调,表明(S)-ZJM-289可通过激活eNOS,释放NO,促进NF-κB-I-κB解偶联,致NF-κB 活化并进入细胞核内进行iNOS基因转录,从而增强IPC效应。

图2 NO经NF-κB通路诱导IPC的分子机制Figure 2 Molecular mechanism of NO-induced IPC via NF-κB singal pathway
4.4 线粒体K+-ATP通道
线粒体K+-ATP通道是脑内产生IPC的重要靶点,而PKC的激活被认为是线粒体K+-ATP通道开放的重要环节[26]。已有研究显示,给IPC模型大鼠在脑缺血前注射K+-ATP通道拮抗剂,可阻断脑缺血后IPC效应,而K+-ATP通道激动剂在海马切片中可诱导类似IPC效应[27];NO则可通过环磷酸鸟苷(cGMP)-PKG-PKC通路选择性激动线粒体K+-ATP通道[28-29]。最新研究表明,硝酰基和巯基等自由基也可激动该通道[30]。机制研究表明,线粒体K+-ATP通道的开放可抑制细胞膜去极化[31],增加线粒体呼吸率(Bajgar等,J Biol Chem,2001),减少ROS产生[32],抑制胞内钙的超载,从而抑制神经元凋亡,产生 IPC 效应(Pain等,Circ Res,2000 年)。
5 结语
NO及其代谢物在细胞保护分子信号级联中起重要作用,可促进IPC效应的产生,对其的深入研究有积极的临床意义。笔者所在课题组研究表明,新型NO供体型化合物(S)-ZJM-289用于IPC大鼠时,可显著性降低脑缺血再灌注后的神经元损伤和脑梗死体积,改善神经学功能,其诱导IPC效应的产生与增强eNOS活性和改善线粒体功能相关,它可通过增加eNOS活性,改善内皮细胞功能,增强脑内缺血区域的微循环,促使ERK磷酸化,导致IPC效应的产生。亚硝酸盐是本课题组最新的研究重点,因为它不仅是一种稳定的NO贮存器,能在缺血后对缺血区域发出类似NO的信号,其具有的氧化还原特性又令它在这个信号通路中的作用具有多样性,而且它还是IPC效应调节器,在治疗脑缺血再灌注损伤方面极具潜在价值。尽管亚硝酸盐介导IPC效应的机制尚不清楚,但现有的研究数据表明,它是缺血反馈的生理调节器,为治疗缺血性疾病的很有潜力的药物。
[1]Gidday J M.Cerebral preconditioning and ischaemic tolerance[J].Nat Rev Neurosci,2006,7(6):437-448.
[2]Zhou L,Zhu D Y.Neuronal nitric oxide synthase:structure,subcellularlocalization,regulation,and clinical implications[J].Nitric Oxide Biol Chem,2009,20(4):223-230.
[3]Hall C N,Garthwaite J.What is the real physiological NO concentration in vivo?[J].Nitric Oxide Biol Chem,2009,21(2):92-103.
[4]Saha R N,Pahan K.Regulation of inducible nitric oxide synthase gene in glial cells[J].Antioxid Redox Sign,2006,8(5/6):929-947.
[5]Pannu R,Singh I.Pharmacological strategies for the regulation of inducible nitric oxide synthase:neurodegenerative versus neuroprotective mechanisms[J].Neurochem Int,2006,49(2):170-182.
[6]Bustamante J,Czerniczyniec A,Lores-Arnaiz S.Brain nitric oxide synthases and mitochondrial function[J].Front Biosci,2007,12:1034-1040.
[7]Jagtap P,Szabo C.Poly(ADP-ribose)polymerase and the therapeutic effects of its inhibitors[J].Nat Rev Drug Discov,2005,4(5):421-440.
[8]Ono K,Suzuki H,Sawada M.Delayed neural damage is induced by iNOS-expressing microglia in a brain injury model[J].Neurosci Lett,2010,473(2):146-150.
[9]Zhao H.The protective effect of ischemic postconditioning against ischemic injury:from the heart to the brain[J].J Neuroimmune Pharm,2007,2(4):313-318.
[10]Kaminskia A,Kascha C,Zhang L,et al.Endothelial nitric oxide synthase mediates protective effects of hypoxic preconditioning in lungs[J].Respir Physiol Neurobiol,2007,155(3):280-285.
[11]Stenzel-Poore M P,Stevens S L,King J S,et al Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes:a speculative synthesis[J].Stroke,2007,38(2):680-685.
[12]Liebelta B,Papapetroub P,Alib A,et al Exercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70(heat shock protein-72)and extracellular-signal-regulated-kinase 1/2 [J].Neuroscience,2010,166(4):1091-1100.
[13]Yuan H J,Zhu X H,Luo Q.Noninvasive delayed limb ischemic preconditioning in rats increases antioxidant activities in cerebral tissue during severe ischemia-reperfusion Injury[J/OL].J Surg Res,2010,in Press.[2011-8-16].http://www.sciencedirect.com/science/article/pii/S0022480410009224.
[14]Dirnagl U,Meisel A.Endogenous neuroprotection:mitochondria as gateways to cerebral preconditioning?[J].Neuropharmacology,2008,55(3):334-344.
[15]Zhang Y,Park T S,Gidday J M.Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation[J].Am J Physiol Heart Circ Physiol,2007,292(6):H2573-H2581.
[16]Zhang Q G,Wang R M,Han D,et al.Preconditioning neuroprotection in global cerebral ischemia involves NMDA receptor-mediated ERK-JNK3 crosstalk[J].Neurosci Res,2009,63(3):205-212.
[17]Simon R,Henshall D,Stoehr S,et al.Endogenous mechanisms of neuroprotection[J].Epilepsia,2007,48(s8):72-73.
[18]Atochin D N,Clark J,Demchenko I T,et al.Rapidcerebral ischemic preconditioningin mice deficient in endothelial and neuronalnitric oxidesynthases[J].Stroke,2003,34(5):1299-1303.
[19]Gustavsson M,Mallard C,Vannucci S J,et al.Vascular response to hypoxic preconditioning in the immature brain[J].J Cereb Blood Flow Metab,2007,27(5):928-938.
[20]Wang L,Traystman R J,Murphy S J.Inhalational anesthetics as preconditioning agents in ischemic brain[J].Curr Opin Pharmacol,2008,8(1):104-110.
[21]Kitano H,Young J M,Cheng J,et al.Gender-specific response to isoflurane preconditioning in focal cerebral ischemia[J].J Cereb Blood Flow Metab,2007,27(7):1377-1386.
[22]Li J,Lang J,Zeng Z,et al.Akt1 gene deletion and stroke[J].J Neurol Sci,2008,269(1/2):105-112.
[23]Sampaio W O,dos Santos R A S,Faria-Silva R,et al.Angiotensin-(1-7)through receptor mas mediates endothelial nitric oxide synthase activation via akt-dependent pathways[J].Hypertension,2007,49(1):185-192.
[24]Nijboer C H,Heijnen C J,Groenendaal F,et al.A dual role of the NF-κB pathway in neonatal hypoxic-ischemic brain damage[J].Stroke,2008,39(9):2578-2586.
[25]Rybnikova E,Gluschenko T,Tulkova E,et al.Preconditioning induces prolonged expression of transcription factors pCREB and NF-κB in the neocortex of rats before and following severe hypobaric hypoxia[J].J Neurochem,2008,106(3):1450-1458.
[26]Dave K R,DeFazio R A,Raval A P,et al.Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon[J].J Neurosci,2008,28(16):4172-4182.
[27]Wang L,Zhu Q L,Wang G Z,et al.The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain[J].Neurosci Lett,2011,491(1):63-67.
[28]Queliconi B B,Wojtovich A P,Nadtochiy S M,et al.Redox regulation of the mitochondrial K(ATP)channel in cardioprotection[J].Biochim Biophys Acta,2010,1813(7):1309-1315.
[29]Prime T A,Blaikie F H,Evans C,et al.A mitochondriatargeted S-nitrosothiol modulates respiration,nitrosates thiols,and protects against ischemia-reperfusion injury[J].ProcNatlAcadSciUSA,2009,106(26):10764-10769.
[30]Lundberg J O,Weitzberg E,Gladwin M T.The nitratenitrite-nitric oxide pathway in physiology and therapeutics[J].Nat Rev Drug Discov,2008,7(2),156-167.
[31]Costa A D,Garlid K D.Intramitochondrial signaling:interactions among mitoKATP,PKCε,ROS,and MPT[J].Am J Physiol Heart Circ Physiol,2008,295(2):H874-H882.
[32]Mayanagi K,Gáspár T,Katakam P V,et al.The mitochondrial K(ATP)channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats[J].J Cereb Blood Flow Metab,2007,27(2):348-355.
