慢病毒载体介导人非小细胞肺癌A549细胞Akt2基因靶向抑制
2011-07-27陈勃江李为民
刘 丹 黄 燚 陈勃江 蒲 蓉 曾 静 王 蕾 李为民
肺癌是一种严重威胁人类健康的恶性肿瘤,其中,约80% ~85%是非小细胞肺癌(non-small cell lung cancer,NSCLC)。尽管肺癌治疗已取得显著的进步,但患者5年生存率仍无明显改善[1]。因此探索肺癌发病机制,寻求新的治疗靶点已成为当前的一项重要任务。
Akt信号通路作为潜在的肿瘤治疗靶点,是目前肿瘤治疗领域的研究热点。Akt是一种丝氨酸/苏氨酸蛋白激酶,作为信号通路的重要枢纽,它通过调节下游靶分子活性,参与多种细胞生物学功能,促进肿瘤的发生发展及血管的发生[2-3]。Akt共有三种亚型,包括Akt1、Akt2、Akt3。其中,Akt2是Akt的重要亚型,在许多肿瘤中都发现Akt2的基因扩增、蛋白有过度表达,然而Akt2在NSCLC中的作用尚存在争议,其介导的特异性信号通路也未阐述清楚,相关研究报道较少[4-5]。本研究旨在采用慢病毒shRNA表达载体介导的RNA干扰技术,在NSCLC细胞株中靶向抑制Akt2基因表达,获得稳定干扰Akt2表达的NSCLC细胞株,为进一步探讨Akt2在肺癌中的作用机制奠定研究基础。
材料与方法
一、实验材料
1.细胞株、菌株及病毒载体:人肺腺癌细胞株(A549),293T细胞及大肠杆菌菌株DH5α均为本实验室培养,慢病毒载体系统购自上海吉凯基因公司。
2.主要试剂:DMEM、胎牛血清、胰蛋白酶及双抗购自美国Hyclone公司,质粒抽提试剂盒购自美国Qiagen公司,Age I及EcoRI内切酶购自美国New England Biolabs公司,蛋白提取试剂盒购自南京凯基生物公司,Akt2抗体购自美国Cell Signaling Technology公司,BCA蛋白定量试剂盒购自美国Pierce公司。
二、实验方法
1.细胞培养:A549及293T细胞以10%胎牛血清的DMEM培养基,100 U/ml青霉素和100 μg/ml链霉素,在37℃、5%CO2、饱和湿度细胞培养箱中进行培养。贴壁生长的细胞2~3 d更换培养液一次,观察细胞贴壁铺满培养皿70%~80%时可进行传代。
2.制备特异性沉默Akt2的重组质粒(pGCsil-shAkt2-GFP载体的构建):根据RNA干扰设计序列原则及既往文献报道,采用Invitrogen公司BLOCK-iT在线设计并进行评估测定(表1),选择最佳动力学参数靶点于上海吉凯基因公司合成含干扰序列的dsDNA oligo(表2)。把合成的引物干粉溶解于退火缓冲液中,经Age I和EcoRI酶切pGCsil-GFP载体,以使其线性化。载体和dsDNA Oligo连接,于16℃连接过夜,干扰序列插入pGCsil-GFP质粒,经感受态细胞5DHα转化、阳性克隆测序鉴定所插入的片段。PCR反应体系:Primer(+):5’-CCTATTTCCCATGATTCCTTCATA-3’;Primer(-):5’-GTAATACGGTTATCCACGCG-3’,pGCsil空载:306bp,阳性克隆:343bp。抽提重组质粒及辅助包装元件载体质粒pHelper 1.0和p-Helper 2.0,通过阳离子脂质体lipofectemin2000介导细胞转染及慢病毒包装,收集浓缩病毒,分装、-80℃保存。

表1 Akt2 siRNA干扰序列设计信息Table 1 Information of Akt2 siRNA interference sequence

表2 干扰序列dsDNA oligo设计信息Table 2 Information of dsDNA oligo interference sequence
3.病毒滴度测定(孔稀释法测定滴度):准备96孔板,以4×104细胞/孔密度铺板,体积为100 μl。37℃、5%CO2孵箱培养24 h,使细胞贴壁。准备7~10个无菌的EP管,倍比稀释病毒原液,并以第一个EP管中的10 μl病毒原液记为1E+1 μl,第二个EP管中为第一个EP管的1/10,记为 1E+0 μl,第三个:1E-1μl…… 第七个:1E-5 μl,第八个:1E-6 μl。吸取96 孔板中待检测细胞孔内的90 μl培养基,丢弃。依次加入90 μl稀释好的病毒溶液。将96孔板置入细胞孵箱培养24 h后,加入完全培养基100 μl,注意不要吹起细胞。继续放入细胞孵箱培养。4 d后,观察荧光表达情况,荧光细胞数随稀释倍数的增加而减少。根据公式:病毒滴度=感染荧光的细胞数/病毒原液量,计算病毒滴度。
4.shAkt2重组慢病毒感染靶细胞A549:将A549细胞按5×104/孔接种于6孔板中,37℃、5%CO2培养箱内培养至细胞融合度约达30%。根据预实验感染条件摸索,A549细胞的MOI值为50,感染条件为完全培养基+5 μg/ml polybrene。根据MOI值加入适量病毒及polybrene,在水平方向轻轻拍打培养板,使培养基和病毒等试剂充分混匀,然后放入孵箱培养孵育。培养24 h后更换为含10%胎牛血清的完全培养基。感染3 d后观察慢病毒报告基因GFP的表达情况。
5.筛选稳定表达shAkt2的细胞克隆:流式细胞分选具有分选精度高(99%以上)速度快的特点。成功感染shAkt2细胞同时表达报告基因GFP蛋白,以GFP为标记获得高纯度稳定转染shAkt2的细胞克隆。根据BD FACSCalibur流式仪操作要求进行分选,将表达GFP强阳性细胞群分选至无菌离心管中。离心管内的细胞悬液平分至培养皿中,37℃,5%CO2孵箱培养细胞。24 h后观察细胞生长状态及GFP表达情况。
6.荧光定量PCR检测细胞Akt2的表达:细胞共分3组:正常对照(A549)组,阴性对照病毒(NC)组,Akt2干扰(shAkt2)组。抽提三组细胞的总RNA,根据Promega公司M-MLV操作说明书进行,总RNA经70℃变性,0℃冰浴退火。经逆转录反应1 h,获得逆转录反应产物-cDNA,取部分用于PCR,余保存在-80℃下。PCR引物信息如下:
Akt2:
上游引物:5’-GCGGAAGGAAGTCATCATTG-3’
下游引物:5’-GTGGGTCTGGAAGGCATAC-3’
设定程序为两步法Real-Time PCR:经变性、退火、延伸过程,并对数据进行Real time分析,分析采用 2-ΔΔCt分析法进行。
7.Western blot检测Akt2的蛋白表达:根据凯基生物蛋白抽提试剂盒操作说明抽提细胞蛋白。并采用二喹啉甲酸(bicinchoninic acid,BCA)法测定总蛋白含量,经SDS-PAGE凝胶电泳,80~100 V电泳1~2 h后,采用湿转方法转膜,用封闭液(含5%脱脂牛奶的TBST溶液),室温、摇床上封闭PVDF膜2 h。4℃孵育过夜。第2天,TBST洗膜5 min×3次。室温孵育二抗(1︰1000)及内参GAPDH(1︰5000)1 h。Millipore公司 Luminata Crescendo Western HRP substrate显色液孵育3 min,Molecular Image ChemiDoc XRS System(Bio-Rad)曝光5 s~10 min。Quantity one分析软件测定条带灰度。
8.统计学分析:用SPSS 13.0统计软件进行分析,数据表示为,采用方差分析(analysis of variance,ANOVA),最小显著差(least-significant difference,LSD)法两两比较。P<0.05有统计学意义。
结 果
一、重组pGCsil-shAkt2-GFP质粒的鉴定
1.阳性克隆的PCR鉴定:pGCsil-shAkt2-GFP连接产物转化感受态大肠杆菌,经37℃过夜培养,挑选出的阳性克隆溶于LB培养基,作为菌落PCR模板。PCR凝胶电泳图如图1,以dd H2O(1泳道)为阴性对照,排除系统中外源核酸污染导致的假阳性结果,Maker指示PCR产物的片段大小(3泳道),结果显示,阳性克隆组(4-7组)PCR产物条带大小约为343bp(从载体中切出24bp),大于空载体自连对照组(2泳道)(片段大小约306bp),说明阳性克隆已插入外源片段,需进一步测序验证。

图1 pGCsil-shAkt2-GFP质粒菌落PCR电泳图Figure 1 PCR electrophoretogram of pGCsil-shAkt2-GFP plasmid
2.重组质粒的测序:挑选重组质粒阳性克隆送至Invitrogen公司测序。测序报告见图2,结果显示:阳性克隆序列内含有干扰Akt2的shRNA片段序列,片段序列为:
CcggGCACAGGTTCTTCCTCAGCTTCAAGAGAGCTGAGGAAGAACCTGTGCTTTTTg,片段大小为57 bp,与之前所设计的针对Akt2的shRNA片段序列完全符合。证明shAkt2序列已成功克隆到慢病毒pGCsil-GFP载体中。

图2 重组pGCsil-shAkt2-GFP质粒测序鉴定Figure 2 Sequence of recombinant pGCsill-shAkt2-GFP plasmid
二、shAkt2慢病毒表达载体的滴度测定
逐孔稀释法测定病毒滴度,病毒原液经倍比稀释感染293T细胞,培养4 d后,观察细胞荧光表达情况。以病毒原液记为1E+1μl,递次稀释十倍,依次类推稀释病毒记为1E+0μl,1E-1μl,1E-2μl……1E-6μl。根据细胞荧光表达情况如图3所示,在加入1E-6μl病毒原液的孔中观察到3个带有荧光的细胞,则本实验获得重组Akt2-shRNA慢病毒滴度为:3/(1E-6)=3E+6 TU/μl,即3E+9 TU/ml,满足实验需要。
三、稳定干扰Akt2基因细胞克隆的筛选
1.重组慢病毒shAkt2感染A549细胞:将重组慢病毒shAkt2和NC对照病毒同时感染人肺腺癌细胞株A549,24 h后更换为完全培养基,观察细胞荧光表达情况。5 d后,A549细胞感染效率达80%以上,感染后细胞状态良好(图4)。

图3 重组shAkt2慢病毒滴度测定Figure 3 Lentivirus titer of recombinant shAkt2

图4 各组细胞病毒感染率及GFP荧光表达水平Figure 4 Virus infection,and GFP expression in cells
2.流式分选筛选稳定干扰Akt2的细胞克隆:成功感染shAkt2细胞同时表达基因GFP蛋白,以GFP为标记,经流式细胞仪分选GFP强阳性细胞,占细胞总数的90.1%(图5),获得稳定沉默Akt2基因的细胞克隆。

图5 流式细胞分选表达GFP强阳性的shAkt2细胞Figure 5 GFP Strong positive shAkz cells obtained by flow cytometer
四、Akt2基因干扰效果的验证
1.荧光定量PCR检测Akt2 mRNA的表达水平:抽提A549、NC及纯化后shAkt2细胞组细胞的总RNA,采用β-actin作为内参,2-ΔΔCt分析法分析Akt2 mRNA含量,方差统计分析结果示,阴性对照组与未感染病毒A549细胞mRNA水平无明显变化,而shAkt2细胞组与A549及NC组比较,Akt2 mRNA明显抑制(P=0.001和 P=0.002,图6),与正常组相比,抑制率约为 63.39%,提示shAkt2细胞克隆显著干扰Akt2 mRNA的表达。
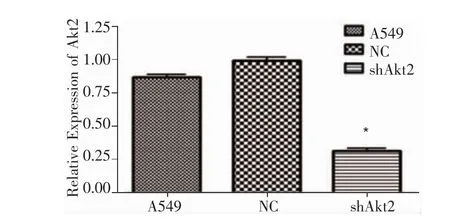
图6 各组细胞中Akt2 mRNA的表达水平Figure 6 Akt2 mRNA expression in A549,NC and shAkt2
2.Western blot检测Akt2的蛋白表达水平:未感染病毒A549细胞、NC阴性对照组和稳定转染shAkt2细胞组抽提总蛋白,以β-actin为内参,进行western blot检测,统计各组条带灰度值,比较Akt2蛋白在各组细胞中的表达水平。如图7所示,shAkt2细胞组中Akt2的蛋白表达较其他两组明显抑制(P<0.01),与正常对照组A549相比,蛋白表达量抑制率为91.56%,说明shAkt2组细胞Akt2蛋白表达获得稳定抑制。
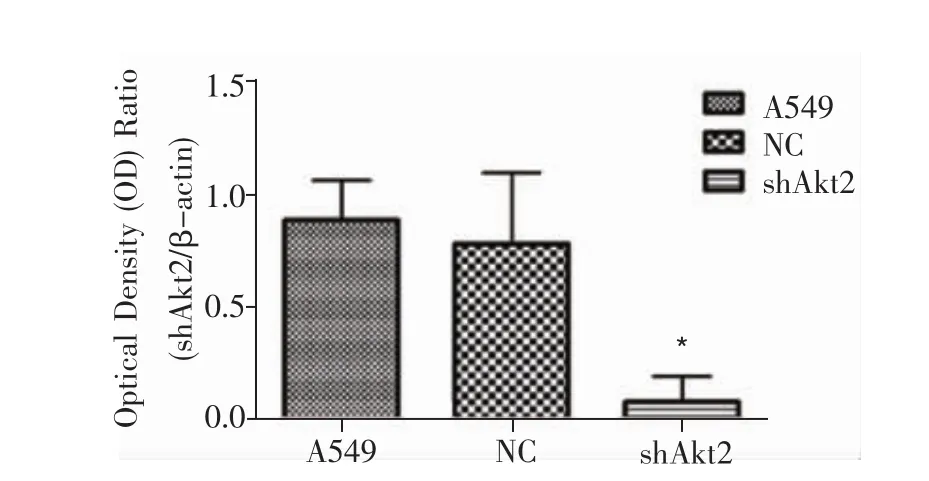
图7 A549、NC及shAkt2组细胞Akt2蛋白表达水平的比较Figure 7 Comparison of Akt2 protein expression in A549,NC and shAkt2
讨 论
Akt2基因是Akt家族的重要亚型,负责编码丝氨酸/苏氨酸蛋白激酶,定位于人类染色体的19q13.1-q13.2。已证实Akt2作为癌基因参与了多种肿瘤的发生和发展,并介导肿瘤细胞生长、黏附、运动、侵袭和转移。本研究拟通过制备有效的靶向干扰Akt2基因的重组慢病毒载体,感染肺腺癌细胞株A549,并检测其对A549细胞的Akt2基因的沉默效率,建立稳定干扰Akt2基因的细胞克隆,为后续研究Akt2在NSCLC中的作用奠定基础。
RNA干扰(RNA interference,RNAi)技术是由双链RNA(double-stranded RNA,dsRNA)诱导同源靶基因mRNA特异性降解,从而特异性抑制目的蛋白的表达,导致转录后基因沉默的现象[6]。由于其具有高度靶向性和高效性等特点,成为肿瘤研究领域的重要方法,广泛应用于基因功能、干细胞发育、信号转导及抗肿瘤治疗等多学科领域。
在细胞内,dsRNA分子被胞内RNA酶Dicer降解为大小约21~23个碱基的小片段RNA,即siRNA,随之,siRNA在RNA解旋酶的作用下解链成正义链和反义链,其中,反义siRNA与体内的内切酶、外切酶、解旋酶等结合形成 RNA诱导的沉默复合物(RNA-induced silencing complex,RISC)[7-8]。RISC具有核酸酶功能,能够与外源性基因表达的mRNA在同源区内进行特异性的结合,并在此结合部位切割降解mRNA,诱发宿主细胞产生针对这些mRNA的降解反应。同时,在RNA聚合酶作用下,以siRNA为引物与靶RNA结合从而合成大量的dsRNA,使RNAi作用进一步放大,最终促使靶mRNA完全降解[6,9-10]。RNAi具有高效、特异的靶向沉默基因的作用,目前已经成为基因治疗和基因功能研究的重要工具。
目前RNAi的制备方法主要包括化学合成、体外转录、体内表达。尽管化学合成的siRNA能特异性抑制细胞内同源基因的表达,但在细胞内容易降解,稳定性差,而将小发卡RNA(small hairpin RNA)导入细胞后,能在细胞内稳定转录生成shRNA,并进一步加工成靶基因特异性siRNA,从而发挥稳定、长期抑制靶基因的表达作用[11-12]。然而,一般的shRNA尽管能够转染到分裂细胞中,但是很难导入到非分裂细胞、原代细胞及人或动物体内去。因此,利用病毒载体包括慢病毒及腺病毒等,因其操作方便、转染效率高及稳定性表达等而被广泛应用。使用腺病毒载体感染细胞时,病毒DNA游离在细胞核内,不能整合到染色体上,在体内不能实现稳定长期表达,并且容易引起免疫反应[13-15]。慢病毒载体是以人类免疫缺陷型病毒(human immunodeficiency virus,HIV)为基础发展起来的基因治疗载体,既能够感染分裂细胞,而且可以感染非分裂细胞,尤其是携带shRNA的慢病毒载体,不仅能够广泛的感染宿主细胞,而且也能够稳定整合于靶细胞的基因组内,具有表达时间长、靶向性佳,不易诱发宿主免疫反应、安全性好等优点。因此,表达shRNA的慢病毒载体已广泛应用于多种细胞内基因功能的研究[16]。
本研究所使用的慢病毒载体系统由pGCsil-LV载体、p-Helper 1.0载体和p-Helper 2.0载体组成。pGC-LV载体中含有HIV的基本元件5’LTR和3’LTR以及其他辅助元件,例如WRE(woodchuck hepatitis virus posttranscriptional regulatory element)。根据不同的实验目的针对pGC-LV载体改造以进行启动子活性研究、基因表达研究、RNAi等研究。pHelper 1.0载体中含有HIV病毒的gag基因,编码病毒主要的结构蛋白;pol基因,编码病毒特异性的酶;rev基因,编码调节gag和pol基因表达的调节因子。pHelper 2.0载体中含有单纯疱疹病毒来源的VSV-G基因,提供病毒包装所需要的包膜蛋白。
本研究根据RNAi干扰原理,针对Akt2设计干扰序列,并合成其寡核苷酸,连接至已线性化的慢病毒pGCsil-LV载体U6启动子下游,在大肠杆菌H5α内同源重组,经过限制性内切酶酶切和测序,证实重组成功后,转入哺乳动物细胞,载体表达出shRNA,这种shRNA很快在细胞中加工成为21~23个碱基大小的dsRNA分子,随后启动RNAi过程。同时质粒携带GFP序列,用于判读转染效果并筛选出稳定转染细胞。经进一步采用荧光定量PCR及western-blot检测Akt2基因表达水平。结果显示,实验组Akt2 mRNA水平较对照组下降大于63.39%,而蛋白水平减少91.56%,说明本研究构建的慢病毒shRNA表达载体在A549细胞内能持续、特异、高效的抑制Akt2基因表达,为后续研究奠定了技术基础,也为肿瘤基因靶向沉默治疗带来了新希望。
1 Jemal A,Siegel R,Ward E,et al,Thun MJ.Cancer statistics,2009[J].CA Cancer J Clin,2009,59(4):225-249.
2 Borders EB,Bivona C,Medina PJ.Mammalian target of rapamycin:biological function and target for novel anticancer agents[J].Am J Health Syst Pharm,2010,67(24):2095-2106.
3 Bjornsti MA,Houghton PJ.The TOR pathway:a target for cancer therapy[J].Nat Rev Cancer,2004,4(5):335-348.
4 Dillon RL,Muller WJ.Distinct biological roles for the akt family in mammary tumor progression[J].Cancer Res,2010,70(11):4260-4264.
5 Stambolic V,Woodgett JR.Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration[J].Trends Cell Biol,2006,16(9):461-466.
6 Stevenson M.Therapeutic potential of RNA interference[J].N Engl J Med,2004,351(17):1772-1777.
7 Milhavet O,Gary DS,Mattson MP.RNA interference in biology and medicine[J].Pharmacol Rev,2003,55(4):629-648.
8 Kim VN.RNA interference in functional genomics and medicine[J].J Korean Med Sci,2003,18(3):309-318.
9 Bernstein E,Caudy AA,et al,Hannon GJ.Role for a bidentate ribonuclease in the initiation step of RNA interference[J].Nature,2001,409(6818):363-366.
10 Nicholson RH,Nicholson AW.Molecular characterization of a mouse cDNA encoding Dicer,a ribonucleaseⅢ ortholog involved in RNA interference[J].Mamm Genome,2002,13(2):67-73.
11 Elbashir SM,Harborth J,Lendeckel W,et al.Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells[J].Nature,2001,411(6836):494-498.
12 秦俊文,谢琪璇,蔡冬青,等.shRNA慢病毒质粒的构建技巧[J].中国生物工程杂志,2011,31(3):124-127.QIN Jun-wen,XIE Qi-xuan,CAI Dong-qing,et al.Framing skills of ShRNA slow virus plasmid[J].J Clin Biotechnol,2011,31(3):124-127.
13 Sandoval Rodriguez AS,Salazar Montes AM,Armendariz-Borunda J.Viral vectors in gene therapy.Advantages of the adenoassociated vectors[J].Rev Gastroenterol Mex,2005,70(2):192-202.
14 Gardlik R,Palffy R,Hodosy J,et al.Vectors and delivery systems in gene therapy[J].Med Sci Monit,2005,11(4):RA110-RA121.
15 Worgall S.A realistic chance for gene therapy in the near future[J].Pediatr Nephrol,2005,20(2):118-124.
16 Cockrell AS,Kafri T.Gene delivery by lentivirus vectors[J].Mol Biotechnol,2007,36(3):184-204.
