环己烷气相氧化脱氢催化剂的研究进展
2011-07-25余国贤李忠铭
晋 梅,余国贤,李忠铭
(江汉大学化学与环境工程学院,湖北 武汉 430056)
1 引言
环己烯是一种重要的有机化工原料,广泛用于医药、食品、农用化学品、饲料、聚酯等精细化工产品的生产。自Sato等[1]从环己烯直接氧化合成己二酸后,环己烯就被认为是合成环己酮、环己醇和己二酸的最佳原料[2,3]。环己烯可通过两种方法获得:一是目前工业上采用的苯选择加氢反应,然而其转化率低、选择性不高、副产物环己烷较多且难以利用[4];二是环己烷脱氢反应,但脱氢生成苯是热力学稳定体系[5]。因此,环己烷气相氧化脱氢制环己烯将成为生产己二酸的新途径,也是充分利用苯部分加氢副产物环己烷以形成环己烷-苯-环己烯循环利用并最大化生产环己烯的绿色工艺路线。
环己烷气相氧化脱氢反应由一系列平行串联反应组成且在较高反应温度下进行,目的产物环己烯为反应的中间产物,存在着环己烷转化率和环己烯选择性之间的矛盾。反应的关键是解决反应过程中转化率和选择性之间的协调问题以及催化剂在高温下的稳定性问题。为了解决这一问题,一个切实可行的方法是采用可活化环己烷分子中C-H键同时避免生成C-O键的催化剂。近年来,国内外针对环己烷氧化脱氢反应催化剂体系的研究主要集中在阳离子沸石催化剂、复合金属氧化物催化剂以及贵金属丝网催化剂等体系,致力于在较缓和的反应条件、较易的催化剂制备方法下获得较高的环己烯产率和较少的COx排放。作者在此对上述三类催化剂体系所遵循的反应机理、催化性能和反应条件进行详细的阐述,同时对金属氧化物催化剂体系中的钒基、钼基以及镍基催化剂的反应活性中心进行进一步探讨,为今后环己烷氧化脱氢反应催化剂的研究提供参考。
2 环己烷气相氧化脱氢反应机理
对不同催化剂体系的氧化脱氢反应机理进行探讨,寻找反应速控步骤,对提高环己烯的选择性和收率具有十分重要的意义。
2.1 自由基反应机理
Alimardanov等[6]采用沸石载体金属氧化物为催化剂,研究环己烷氧化脱氢反应机理,如式(1)~(9)所示。首先,环己烷在催化剂表面生成环己基自由基,继而脱氢生成环己烯,同时伴随生成小分子烃及COx的副反应。
(1)
(2)
C6H12+0.5O2→C6H10+H2O
(3)
C6H10+O2→C6H6+2H2O
(4)
C6H10+1.5O2→C5H8+CO2+H2O
(5)
C6H10+2.5O2→C4H8+2CO2+H2O
(6)
C6H10+4O2→C3H6+3CO2+2H2O
(7)
C6H10+5.5O2→C2H4+4CO2+3H2O
(8)
C6H10→C5H7CH3
(9)
2.2 氧化还原反应机理
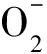
遵循Mars-van Krevelen反应机理的氧化物有V2O5、MoO3、Bi2O3以及Bi2O3-MoO3等,具有反应温度较高、环己烯选择性高的特点。反应机理为:(1)气相环己烷分子和氧气分别在催化剂表面进行吸附;(2)吸附于催化剂表面的气相烃分子与高价态金属氧化物催化剂表面上的晶格氧相互作用,烃分子被氧化为环己基自由基;晶格氧参与反应后,催化剂上的金属氧化物被还原为较低价态;(3)气相氧将低价态的金属氧化物氧化到初始高价态,补充晶格氧,完成催化剂上金属阳离子的氧化-还原循环。反应速控步骤为吸附的环己烷分子中-H的提取。
2.3 均相气相反应和非均相气相反应共存的反应机理
O′Connor等[11,12]在贵金属的单一金属丝网反应器中对环己烷氧化脱氢反应进行研究,提出反应机理,如图1所示。
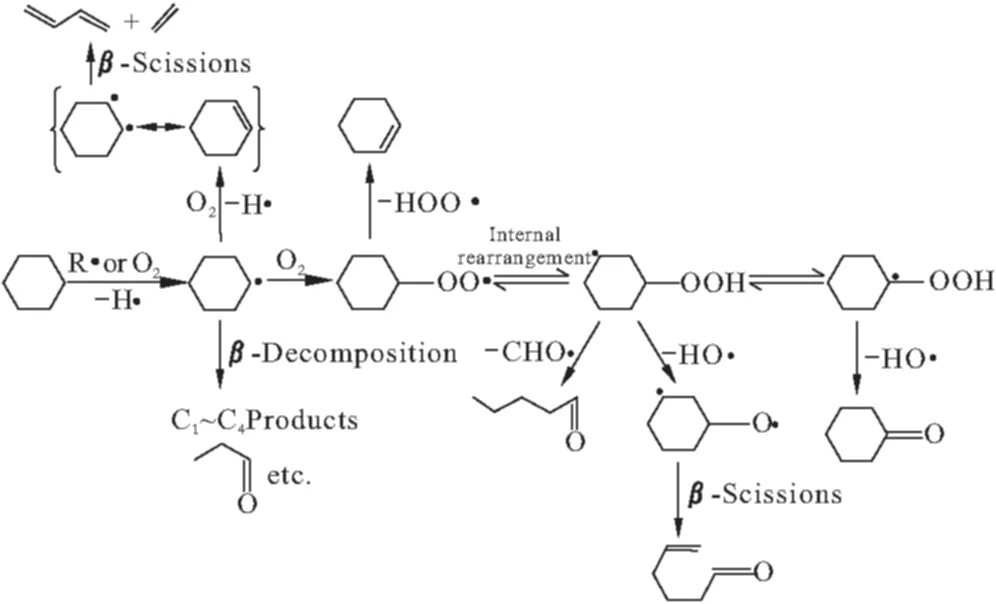
图1 环己烷部分氧化的反应路径
环己烷分子首先进行非均相气相反应生成环己基自由基,而后进一步脱氢生成环己烯。整个反应体系过程在高温及毫秒级接触时间下进行,其中还伴随有均相气相反应,产物中有大量氧化副产物,环己烯分离极其困难。
3 环己烷氧化脱氢制备环己烯的催化剂性能
不同催化剂体系在环己烷氧化脱氢反应中遵循不同的反应机理,因而在环己烷氧化脱氢的反应体系中采用的操作条件和催化性能有较大差异,如表1所示。
从表1可知:(1)环己烷氧化脱氢反应的氧化剂是低浓度的氧气或空气,不仅具有适宜的氧化性且价廉易得;(2)环己烷氧化脱氢反应的温度主要集中在400~600℃;(3)贵金属丝网催化剂的反应的条件最为苛刻,反应温度高达900°C、接触时间<5 ms;(4)采用复合金属氧化物作为催化剂进行环己烷氧化脱氢反应的条件相对较为温和且催化性能相对较好,针对该体系的研究也最多。
4 金属氧化物催化剂催化活性组分
环己烷氧化脱氢反应中,研究最多的金属氧化物催化剂体系主要集中在钒基、钼基和镍基氧化物。
4.1 钒基催化剂
虽然V5+具有很好的活化烃分子上C-H键的能力[29,30],但其结构中V=O有利于C-O键的生成,产物中COx量较多。为了提高目的产物环己烯的产率和选择性,通常向V2O5中加入另一种金属阳离子以形成金属钒酸盐。钒酸盐结构中的V-O-Me键会改变V-O活性中心的电子云密度分布、催化剂表面的酸碱性和活性物种的氧化还原性速率。比较典型的金属阳离子有两种:一种电负性小于钒,如Mg;另一种电负性大于钒,如P。
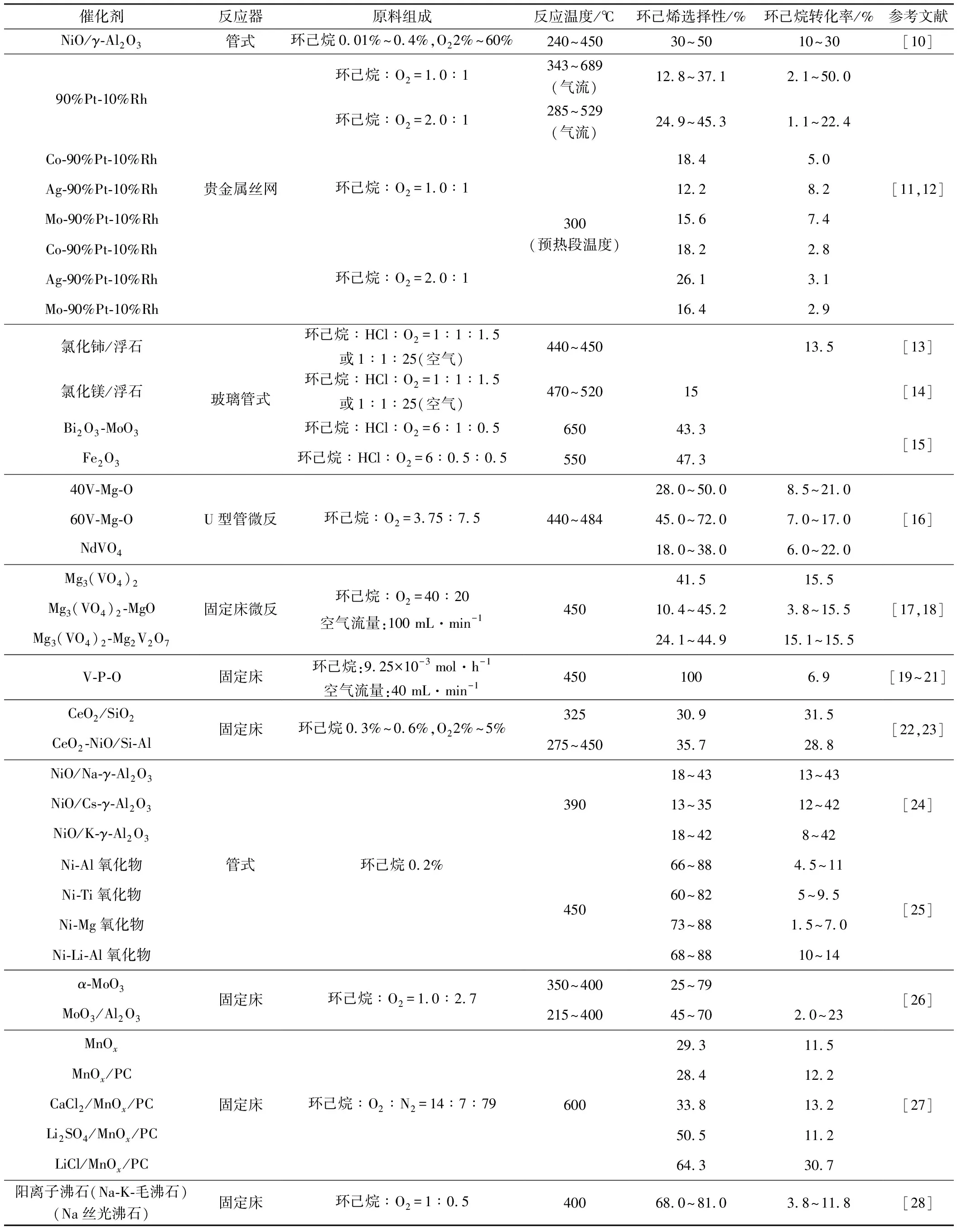
表1 环己烷氧化脱氢反应催化剂的性能
根据V/Mg原子比的不同,V-Mg-O催化剂主要以Mg3(VO4)2、Mg2V2O7与MgV2O6三种稳定形式存在。在环己烷氧化脱氢反应中,Kung等[16]采用共沉淀法制备40(60)V-Mg-O催化剂[Mg3(VO4)2+MgO+V2O5],虽取得较好催化效果,但未对催化活性相进行研究。Grasselli等提出Mg3(VO4)2晶相具有“孤立活性位”、适量的晶格氧含量、催化剂表面弱碱性以及较低的金属离子可还原性,可作为环己烷氧化脱氢反应的催化活性相[31~33]。方智敏等[34,35]对V-Mg-O进行原位Raman光谱研究表明:V4+⟺V5+氧化还原对易于形成氧缺位而有利于高温吸附并活化氧分子,转化为晶格氧以参与环己烷氧化脱氢反应,且Mg3(VO4)2、Mg2V2O7和MgV2O6的表面活性位为V4+-O-Mg-O-V5+、V4+-O-V5+和V5+-O。其中V4+-O四面体配位结构是氧化脱氢反应的活性位,有利于环己烯选择性的提高;V5+-O八面体配位结构是加氧氧化反应的活性位[36]。
在Carrazán等[37]和Gao等[38]研究的基础上,Jin等[17]对催化活性相与其它晶相间的协同效应进行了研究。他们认为,在Mg3(VO4)2活性相中加入适量的MgO时,固溶的MgO不仅占据环己烷氧化脱氢反应的催化活性位且增强了催化剂的表面碱性;当加入Mg2V2O7时,晶相间由于溢流氧产生的遥控机理作用而促进溢流氧从供体迁移到受体上并与受体作用以修饰催化活性位。类似地,Pantazidis等[39]认为过量的MgO不仅可调节催化剂的氧化还原性能,同时可改变催化剂表面酸碱性,并提出适度增强催化剂的碱性有利于V4+的生成、不利于表面碳酸盐的生成。
P的电负性大于钒,因此催化剂表面亲核性变差而导致催化剂表面呈现弱酸性。与V-Mg-O催化剂相同,V-P-O晶相结构中由于PO4基团与VO6基团共享的氧所处结构不同[40],通常以三个稳定的晶相结构形式存在:αI-VOPO4、αⅡ-VOPO4和β-VOPO4。Herrmann等[41]研究V-P-O催化剂导电性发现,V-P-O属于p型半导体材料,主要电荷载体为正电荷空穴P+,且若要达到最佳催化效果必须有V5+存在。活性位的氧化-还原通过(VO)3+⟺(VO)2++P+和P++O2-⟺O-来完成,参与反应的氧物种为晶格氧物种,这一结论在Abon等[42]用18O2同位素进行的氧化脱氢反应实验中得到了验证。朱宇君等[19~21]仅采用αⅠ-VOPO4为环己烷氧化脱氢反应催化剂时,产物中并没有出现环己烯,究其原因主要为:一方面αⅠ-VOPO4呈弱酸性,不利于中间产物环己烯的脱附;另一方面,V5+的存在可使得催化剂达到最佳催化效果,但又利于深度氧化反应的进行。为了提高环己烯的选择性,他们采用醋酸共进料的方法,通过醋酸分子的-OH与αⅠ-VOPO4晶相中V=O形成氢键吸附在催化剂表面,形成孤立的活性位点。
4.2 钼基催化剂
MoO3为八面体结构的酸性、非严格化学计量氧化物,其内部结构存在大量氧空穴,结构缺陷和气相氧之间通过Mo5+⟺Mo6+氧化还原偶保持动态平衡。其中Mo6+八面体结构利于深度氧化反应,Mo5+不饱和配位结构则有利于烯烃选择性的提高。Cadus等[43]对Mg-Mo-O催化剂进行EPR表征,发现较高Mo5+含量的催化剂在氧化脱氢反应中具有好的催化性能且MoO3稍过量利于Mo5+物种的形成。同时,Allison等[44]提出了含有相邻表面双氧位的催化活性位的反应活化能低于单一钼双氧位的热力学和量子力学结果,利于烯烃选择性的提高。Alyea等[26]采用金属氧化物气相合成方法制备出含双重二氧钼活性位的α-MoO3及MoO3/Al2O3催化剂用于环己烷氧化脱氢反应,主要的催化活性位为Mo5+物种。
4.3 镍基催化剂
5 结语
环己烷氧化脱氢反应催化剂研究虽取得了一定的进展,但仍然存在很多亟待解决的问题,如催化剂活性与选择性的协调、氧物种类型的选择、工业应用等。因此,开发新型催化剂体系、新的环己烷氧化脱氢反应途径势在必行。工艺应用研究、反应器的设计以及反应器中传热传质问题的研究还需进一步加强。
总之,环己烷氧化脱氢制取环己烯不仅为环己烯的合成开辟了一条新的途径,且为高效利用来源丰富、价格便宜的环己烷提供了新的思路,具有广阔的工业应用前景。
[1]Sato K,Aoki M,Noyori R.A "green" route to adipic acid:Direct oxidation of cyclohexenes with 30 percent hydrogen peroxide [J].Science,1998,281(11):1646-1648.
[2]Schuchardt U,Cardoso D,Sercheli R,et a1.Cyclohexane oxidation continues to be a challenge[J].Appl Catal A:General,2001,211(1):1-17.
[3]宫红,姜恒,吕振波.己二酸绿色合成新途径[J].高等学校化学学报,2000,21(7):1121-1123.
[4]Liu S C,Liu Z Y,Wang Z,et al.Characterization and study on performance of the Ru-La-B/ZrO2amorphous alloy catalysts for benzene selective hydrogenation to cyclohexene under pilot conditions[J].J Chem Eng,2008,139(1):157-164.
[5]Riad M,Mikhail S.Dehydrogenation of cyclohexane over molybdenum/mixed oxide catalysts[J].Catal Comm,2008,9(6):1398-1403.
[6]Alimardanov K M.Kineties and mechanism of cyelohexane dehydrogenation to cyclohexene in the presence of molecular oxygen[J].J Petroleum Chemistry:USSR (English Translation of Neftekhimiya),1991,31(1):22-29.
[7]Bielan′ski A,Haber J.Oxygen in catalysis on transition metal oxides[J].Catal Rev Sci Eng,1979,19(1):1-41.
[8]辛勤.固体催化剂研究方法[M].北京:科学出版社,2004:245-250.
[9]Sokolovskii V,Arena F,Giordano N,et al.Role of acid-base properties of SiO2-based catalysts in the selective oxidation of propane[J].J Catal,1997,167(1):296-299.
[10]Patcas F,Pateas F C.Reaction pathways and kinetics of the gas-phase oxidation of cyclohexane on NiO/γ-Al2O3catalyst[J].Catal Today,2006,117(1):253-258.
[11]O′Connor R P,Klein E J,Henning D,et al.Tuning millisecond chemical reactors for the catalytic partial oxidation of cyclohexane[J].Appl Catal A:General,2003,238(1):29-40.
[12]O′Connor R P,Schmidt L D.Catalytic partial oxidation of cyclohexane in a single-gauze reactor[J].J Catal,2000,191(1):245-256.
[13]Tallman R C,Ark E D.Conversion of cyclohexane to cyclohexene[P].USP 3 108 144,1963-10-22.
[14]Boonton N J,Morristown N J.Producing cyclohexene or alkycyclohexene[P].USP 4 356 337,1982-10-26.
[15]赵红坤,雒廷亮,杨杰,等.环己烷氧化脱氢制备环己烯的研究现状及展望[J].浙江化工,2002,33(4):51-54.
[16]Kung H H,Kung M C.Oxidative dehydrogenation of alkanes over vanadium-magnesium oxides[J].Appl Catal A:General,1997,157(1-2):105-116.
[17]Jin M,Cheng Z M.Oxidative dehydrogenation of cyclohexane to cyclohexene over Mg-V-O catalysts[J].Catal Lett,2009,131(3-4):266-278.
[18]Jin M,Cheng Z M,Gao Y L,et al.Oxidative dehydrogenation of cyclohexane with Mg3(VO4)2synthesized by the citrate process[J].Mater Lett,2009,63(23):2055-2058.
[19]朱宇君,李静,杨向光,等.醋酸对环己烷气相氧化脱氢产物选择性影响的研究[J].高等学校化学学报,2006,27(6):1118-1120.
[20]Zhu Y J,Li J,Yang X G,et al.A new route to control product selectivity in the oxidative dehydrogenation of cyclohexane and cyclohexene[J].Chem Lett,2004,33(7):822-823.
[21]Zhu Y J,Li J,Xiao F X,et al.Effect of different VOPO4phase catalysts on oxidative dehydrogenation of cyclohexane to cyclohexene in acetic acid[J].J Mole Catal A:Chemical,2006,246(1):185-189.
[22]Kubo Toshihiko.Oxidative dehydrogenation of cyclohexane to cyclohexene over zeolite catalyst[J].Nippon Kagaku Kaishi,1973,12:2257-2263.
[23]Hayakawa Takshi.Oxidative dehydrogenation of cyclohexane with Ce catalysts supported on Si[J].J Sekiyu Gakkaishi,1987,30:161-165.
[24]Patcas F,Honicke D.Effect of alkali doping on catalytic properities of alumina-supported nickel oxide in the selective oxidehydrogenation of cyclohexane[J].Catal Comm,2005,6(1):23-27.
[25]Patcas F,Krysmann W,Honicke D,et al.Preparation of structured egg-shell catalysts for selective oxidations by the ANOF technique[J].Catal Today,2001,69(2):379-383.
[26]Alyea E C,Keane1 M A.The oxidative dehydrogenation of cyclohexane and cyclohexene over unsupported and supported molybdena catalysts prepared by metal oxide vapor deposition[J].J Catal,1996,164(1):28-35.
[27]Zhu H,Ge Q J,Li W Z,et al.Study of Mn-based catalysts for oxidative dehydrogenation of cyclohexane to cyclohexene[J].Catal Lett,2005,105(1):29-34.
[28]Dietz I A G,Carlsson A F,Schmidt L D.Partial oxidation of C5and C6alkanes over monolith catalysts at short contact times[J].J Catal,1996,167(2):459-473.
[29]Hutchings G J,Desmartin-Chomel A,Olier J C,et al.Role of the product in the transformation of a catalyst to its active state[J].Nature,1994,368(1):41-42.
[30]Kung H H,Birkeland K,Bethke G K,et al.The kinetic significance of V5+inn-butane oxidation catalyzed by vanadium phosphates[J].Science,1997,275(1):191-193.
[31]Charr M A,Patel D,Kung M C,et al.Selective oxidative dehydrogenation of butane over V-Mg-O catalysts[J].J Catal,1987,105(2):483-498.
[32]Grasselli R K.Fundamental principles of selective heterogeneous oxidation catalysis[J].Topics in Catal,2002,21(1):79-88.
[33]Patel D,Kung M C,Kung H H.Proceeding,9th international congress on catalysis[C].Calgary:Chem Institute of Canada,Ottawa,1988,4:1553-1555.
[34]方智敏,翁维正,万惠霖.VMgO催化剂上丙烷氧化脱氢反应的原位Raman谱学研究[J].分子催化,1998,12(3):207-213.
[35]方智敏,翁维正,万惠霖.丙烷氧化脱氢VMgO催化剂活性位的研究[J].厦门大学学报(自然科学版),1998,37(4):525-531.
[36]张伟德,沙开清,李基涛,等.丙烷氧化脱氢催化剂V2O5/MPO4(M=A1,Zr,Ca)的研究[J].高等学校化学学报,1999,20(4):608-611.
[37]Carrazán S R G,Peres C,Bernard J P,et al.Catalytic synergy in the oxidative dehydrogenation of propane over MgVO catalysts[J].J Catal,1996,158(2):452-476.
[38]Gao X T,Ruiz P,Xin Q,et al.Effect of coexistence of magnesium vanadate phases in the selective oxidation of propane to propene[J].J Catal,1994,148(1):56-67.
[39]Pantazidis A,Auroux A,Herrmann J M.Role of acid-base redox and structural properties of VMgO catalysts in the oxidative dehydrogenation of propane[J].Catal Today,1996,32(1):81-88.
[40]Volta J C,Olier R,Roullet M,et al.Study byinsitulaser Raman spectroscopy of a VPO catalyst in the course ofn-butane oxidation to maleic anhydride[J].Studies in Surface Science and Catalysis,1993,75:1531-1534.
[41]Herrmann J M,Vernoux P,Béré K E,et al.Insitustudy of redox and of p-type semiconducting properties of vanadyl pyrophosphate and of V-P-O catalysts during the partial oxidation ofn-butane to maleic anhydride[J].J Catal,1997,167(1):106-117.
[42]Abon M,Béré K E,Delichére P.Nature of active oxygen in then-butane selective oxidation over well-defined V-P-O catalysts:An oxygen isotopic labelling study[J].Catal Today,1997,33(1):15-23.
[43]Cadus L E,Gomez M F,Abello M C.Synergy effects in the oxidative dehydrogenation of propane over Mg-MoO4-MoO3catalysts[J].Catal Lett,1997,43(1):229-233.
[44]Allison J N,Goddard W A.Oxidative dehydrogenation of methanol to formaldehyde[J].J Catal,1985,92(1):127-135.
