Peroxisome proliferator-activated receptor gamma inhibits hepatic fibrosis in rats
2011-07-07ZhengWangJiaPengXuYongChaoZhengWeiChenYongWeiSunZhiYongWuandMengLuo
Zheng Wang, Jia-Peng Xu, Yong-Chao Zheng, Wei Chen, Yong-Wei Sun, Zhi-Yong Wu and Meng Luo
Shanghai, China
Peroxisome proliferator-activated receptor gamma inhibits hepatic fibrosis in rats
Zheng Wang, Jia-Peng Xu, Yong-Chao Zheng, Wei Chen, Yong-Wei Sun, Zhi-Yong Wu and Meng Luo
Shanghai, China
BACKGROUND:Hepatic fibrosis is a necessary step in the development of hepatic cirrhosis. In this study we used lentiviral vector-mediated transfection technology to evaluate the effect of peroxisome proliferator-activated receptor gamma (PPAR-γ) on rat hepatic fibrosis.
METHODS:Hepatic fibrosis in rats was induced by CCl4for 2 weeks (early fibrosis) and 8 weeks (sustained fibrosis). The rats were randomly divided into four groups: normal control, fibrosis, blank vector, and PPAR-γ. They were infected with the recombinant lentiviral expression vector carrying the rat PPAR-γgene by portal vein injection. The liver of the rats was examined histologically and hydroxyproline was assessed.In vitroprimary hepatic stellate cells (HSCs) were infected with the recombinant lentiviral expression vector carrying the rat PPAR-γgene. The status of HSC proliferation was measured by the MTT assay. The protein levels of PPAR-γ,α-smooth muscle actin (α-SMA) and typeicollagen expression were evaluated by the Western blotting method.
RESULTS:In vitrostudies revealed that expression of PPAR-γinhibited expression ofα-SMA and typeicollagen in activated HSCs (P<0.01) as well as HSC proliferation (P<0.01).In vivoexperiments indicated that in the early hepatic fibrosis group, the hydroxyproline content and the level of collageniprotein in the liver in the PPAR-γtransfected group were not significantly different compared to the hepatic fibrosis group and the blank vector group; whereas the expressions of PPAR-γandα-SMA were different compared to the hepatic fibrosis group (P<0.01). In the sustained hepatic fibrosis group, there were significant differences in the hydroxyproline content and the expression of PPAR-γ,α-SMA, and typeicollagen between each group.
CONCLUSION:PPAR-γcan inhibit HSC proliferation and hepatic fibrosis, and suppressα-SMA and typeicollagen expression.
(Hepatobiliary Pancreat Dis Int 2011; 10: 64-71)
peroxisome proliferator-activated receptor gamma; hepatic fibrosis; hepatic stellate cells; lentiviral vector
Introduction
Hepatic fibrosis is caused by a variety of etiological factors, such as viral hepatitis, autoimmune diseases and excessive alcohol intake, which induce abnormal increases and excessive deposition of hepatic extracellular matrix. Hepatic fibrosis is a necessary step in the development of hepatic cirrhosis. Therefore, effective treatment of post-hepatitic cirrhosis, portal hypertension, and other chronic liver diseases depend on the prevention and treatment of fibrosis. Activation of hepatic stellate cells (HSCs) is the key for liver collagen production and fibrosis formation.[1]HSCs, also known as vitamin A-storing cells, lipocytes, interstitial cells, fat-storing cells, or Ito cells, are located in the spaces between the liver sinusoidal endothelial cells and liver parenchymal cells in the hepatic lobule. The fat droplets in the cytoplasm of these cells store 80% of the retinoic acid of the human body in the form of palmitic acid retinaldehyde. In physiological conditions, HSCs play a key role in regulating the dynamic balance of retinoic acid in the body.[2]The most evident changes in HSCs are disappearance of the vitamin A lipid droplet and expression of the hepatic fibrosis marker α-smooth muscle actin (α-SMA), inducing HSCs to express myofibroblast-like traits. Myofibroblasts produce large amounts of extracellular matrix protein, particularly typeicollagen, leading to the development of fibrosis. Inhibition of HSC activation and proliferation is of great significance for the prevention and treatment of hepatic fibrosis and is an important research area.[3]
Peroxisome proliferator-activated receptor (PPAR) is a novel steroid hormone receptor in the liganddependent transcription factor superfamily. PPAR can be activated by fatty acids, fatty acid metabolites and fatty acid-like chemicals. The activated PPAR (α, γ and δ/β) can bind the PPAR response element (PPRE) in the gene regulatory region to activate the corresponding gene's transcription. Therefore, PPAR plays an important role in cell growth, differentiation, and a variety of biological processes. PPAR-γ is expressed in many cells including endothelial cells, vascular smooth muscle cells, monocytes, macrophages and HSCs and plays a key role in fat formation. The activated PPAR-γ can not only bind PPRE, but may also bind retinoic X receptor-α to form heterodimers that play transcriptional regulating roles. Some studies[4,5]showed that HSC activation significantly reduces the expression of PPAR-γ and that PPAR-γ agonists inhibit HSC activation, resulting in reduced expression of α-SMA and collagen, as well as reduced cell proliferation and development of hepatic fibrosis. Increased PPAR-γ expression reduces the synthesis of HSC DNA and results in the decreased expression of α-SMA, collagen and the transforming growth factor-β1. Its expression also transforms HSCS back to a quiescent state, and the storage capacity of retinol palmitate fat is restored. At the same time, PPAR-γ is also involved in the apoptosis of HSCs through a variety of mechanisms.[4-9]
In this study, PPAR-γ lentiviral vector transfection technology was applied to induce HSC activation in vitro and overexpress PPAR-γ in an in vivo rat model of hepatic fibrosis. We determined the degree of fibrosis in rat liver, proliferation of activated HSCs, and expression of α-SMA, typeicollagen and hydroxyproline to investigate the role of increased PPAR-γ expression in the activation and proliferation of HSCs and determine whether increased PPAR-γ expression could inhibit hepatic fibrosis in rats.
Methods
Experimental animals and reagents
Male Sprague-Dawley rats (weighing 350-400 g) were purchased from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences and housed in the Animal Laboratory of Shanghai Jiaotong University School of Medicine. The animals were given standard pellet food and water ad libitum and subjected to a 12-hour light/ 12-hour dark cycle. The room temperature was 18-20 ℃, and the humidity was 60%-70%. All the animal experiments were performed in the Animal Experiment Scientific Centre of Shanghai Jiaotong University School of Medicine. The study was approved and performed according to the Laboratory Animal Care and Usage Manual.
The following reagents were used in this study: RPMI-1640 cell culture medium powder (Gibco, USA), type IV collagenase (Sigma, USA), PPAR-γ gene clone and pGC-FU-3FLAG vector (Genechem, China), antigoat PPAR-γ antibody (Lifespan, USA), anti-rabbit α-SMA (Abcam, UK), and anti-rabbit typeicollagen polyclonal antibody (Boster, China).
Construction and identification of the PPAR-γexpression plasmid
The primers used to amplify full-length PPAR-γ were synthesized by Shanghai Genechem (China), and the sequences were:
a) PPAR-γ-AgeI-F 5'-GAT GAT GAC GAC AAA CCG GTC ATG GGT GAA ACT CTG GGA GAT CCT CCT GTT GAC CCT GAG C-3. This primer contains the complement bases, the restriction sites for Agei(the T bases were removed to adjust the reading frame) and a partial sequence of the 3' end of the target gene.
b) PPAR-γ-M-R 5'-CTT GTG AAG TGC TCA TAG GCA GTG CAT CAG CGA AGG CAC CAT GCT CAG GGT CAA CAG GAG-3. This primer was used to correct the template sequence with the N-terminal deletion.
c) PPAR-γ-M-F 5'-CCT ATG AGC ACT TCA CAA GAA ATT ACC ATG GTT GAC ACA GAG ATG CCA TTC TGG CCC AC-3. This primer was used to correct the template sequence with the N-terminal deletion and amplify the 5' end of the target gene.
d) PPAR-γ-SEQF 5'-GGA TTC ATG ACC AGG GAG-3. This primer was located in the coding sequence of the target gene and used for the identification of the transformant by colony PCR.
e) EGFP-N-R 5'-CGT CGC CGT CCA GCT CGA CCA G-3'. This primer was located in the N terminal of the enhanced green fluorecence protein (EGFP) gene and used for the identification of the transformant by colony PCR and sequencing. The coding region was amplified by PCR.
Each of these fragments was amplified and ligated into the pGC-FU-3FLAG vector, linearized with Age I, and incubated for 15 minutes at 23 ℃ and 42 ℃. The recombinant was transformed into fresh competent E. coli cells prepared by the calcium chloride method (Experiment Guide of Molecular Cloning, 2nd edition, page 55) and incubated for 16 hours at 37 ℃. Thirty cycles of denaturation (94 ℃ for 30 seconds), annealing (60 ℃for 30 seconds), and extension (72 ℃ for 30 seconds) were completed. The positive clones were confirmed by sequencing prior to transfection in mammalian cells. The plasmids were transfected into 293T cells, and theexpression of the confluency protein was observed under a fluorescence microscope. Western blotting was also used to detect the expression of the confluency protein. The virus titer was determined by quantitative real-time PCR.

Table. Groups of Sprague-Dawley rats
Generation of a rat model of hepatic fibrosis
The rat groupings are shown in the Table. The rats were given intraperitoneal injections of 50% CCl4(CCl4∶vegetable oil=1∶1) at a dose of 0.15 ml/100 g body weight and were also given oral 10% ethy1 alcohol in groups F (hepatic fibrosis), N (blank vector) and P (PPAR-γ-transfected). The group C (normal control) was given the same dose of vegetable oil instead of CCl4by intraperitoneal injection. Injections were given twice a week for 2 weeks or 8 weeks, according to the grouping. One week after CCl4/ethanol/control treatment, the rats received an intravenous injection according to their subgrouping. The groups C and F were injected with the virus-free dilution solution only. The rats in the group N were injected with 0.5 ml of blanking plasmid virus solution (5×107TU/ml). The rats in the group P were injected with 0.5 ml of PPAR-γ virus solution (Ringer's solution, 5×107TU/ml). The injection time was 7 seconds in all groups. The rats were sacrificed 2 weeks after injection, and the liver was obtained for further assessments.
HSC separation and culture
The liver nonparenchymal cells were separated by collagenase perfusion through the portal vein, and the primary HSCs were obtained by Nycodenz gradient centrifugation.[10,11]The morphology and growth of HSCs were observed under an inverted fluorescence microscope. After the first replacement of culture medium, the HSCs were washed once with PBS and observed immediately at 328 nm excitation wavelength. After 48 hours of culture, the HSCs were identified using ED2 immunohistochemical staining.
The HSCs were stimulated with platelet-derived growth factor, and the activated HSCs were divided into 3 groups: control (C), lentiviral vector control group (NC), and PPAR-γ-overexpression group (PPAR-γ). The transfection of cells was performed using an MOI of 100. HSCs were cultured separately in 6-well plates 7 days prior to infection. After transfection for 3 days, the lentiviral transfection efficiency was observed under the fluorescence microscope. RT-PCR, Western blotting and MTT were performed 5 days after transfection.
Hepatic histology and grading of liver fibrosis
Hepatic tissue was fixed in 10% formalin and embedded in paraffin. The paraffin-embedded tissue was sectioned 5 μm thick, placed on slides, deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. Conventional hematoxylin and eosin staining was done. After HE staining, the sections were observed under a light microscope by three pathologists.
Hepatic fibrosis is divided into five stages according to the standards developed by the Hepatic Fibrosis Group of the Chinese Institute of Liver Diseases: S0, no fibrosis; S1, enlargement of fibrosis in the portal areas, fibrosis localized in the sinus surroundings and lobule; S2, fibrosis in the region around the portal area or the appearance of a fiber gap with an intact lobule structure; S3, formation of numerous fiber gaps with lobule structure disorder but no cirrhosis; and S4, early stage of cirrhosis. The liver biopsy was taken, graded and scored by a semi-quantitative scoring system (SSS).[12]SSS is a mathematical model, representing the severity of disease.
Detection of hydroxyproline in liver tissue
One hundred milligrams of wet liver tissue was taken after the rats were sacrificed, and 2.5 ml doubledistilled water was added. The tissue was homogenized at 4 ℃ (1800 rpm, 50 seconds). A 100-μl sample was taken to determine the total protein concentration using the biuret method, and the remaining sample was lysed for 18 minutes at 100 ℃ by adding an equal volume of 12 mol/L HCl. The mixture was then filtered. Then, 100 μl of lysed sample was dried to make 0.2-1.6 μg of standard hydroxyproline; this sample was maintained as a control. Each sample and the standard control (two replicates) were reacted using chloramine-T solution and Ehrlich's solution for 30 minutes at 50 ℃ before the absorbance (A) was measured at 558 nm by a spectrophotometer. The regression equation of the standard curve was generated to calculate the concentrations of samples, which were then corrected by the amount of total protein. The content of hydroxyproline in the total protein of the liver tissue was represented as μg/g.
Detection of protein expression of PPAR-γ,α-SMA and typeicollagen in HSCs and liver tissue by Western blotting
The supernatants were aspirated and the cells were washed twice in PBS. The proper amount of pre-cooled2× lysis buffer was added. The cells were deplated, transferred to a tube, and then lysed on ice for 15 minutes. The protein concentration was determined and adjusted to 2 μg/μl. Next, 2× loading buffer was added to each sample and boiled for 5-10 minutes. Forty micrograms of total protein was loaded into each well of a 10% SDS-polyacrylamide gel, electrophoresed at 30 mA for 2 minutes, and then transferred to a polyvinylidene fluoride membrane at 400 mA for 2 hours. The membrane was blocked in 5% milk in Trisbuffered saline (TBS) for 1 hour at room temperature. The primary antibody was added and incubated with the membrane for 2 hours at room temperature, and then washed three times with TBS/Tween 20. A secondary antibody was added to the membrane and was incubated at room temperature with gentle agitation. Two hours later, the membrane was washed three times with TBS/Tween 20 for 10 minutes per wash. The bands were visualized using an enhanced chemiluminescence kit after exposure to X-ray film.
Statistical analysis
Data were expressed as mean±SD. They were analyzed using SPSS software version 12.0 (SPSS Inc., Chicago, IL, USA). Student's t test was used to process the data. A P value less than 0.05 was considered statistically significant.
Results
HSC separation and culture
The yield of HSCs was 2.5-3.2×107/liver, the average viability was 98.0±1.8%, and the average purity was 94.0±1.7%. Inverted microscope observation showed that cells began to adhere 30-60 minutes after plating. The cells were fusiform 72 hours later and reached an 80%-90% confluence after 5-7 days of culture. The cells displayed green fluorescence under 325-nm UV light. The immunohistochemistry showed that desmin protein was expressed in both the static and activated HSCs; however, α-SMA protein was not expressed in the static HSCs. Desmin and α-SMA protein were both expressed in the cytoplasm of activated HSCs.
Construction of pGC-FU-3FLAG-PPAR-γ
A 475-bp fragment was amplified by PCR in the positive clone, which was in line with the expected size. The sequencing result was identical to the known PPAR-γ sequence in GenBank. GFP expression in HSCs was detected by fluorescence microscopy 24 hours after transfection. Western blotting showed a 90-kD band in HSC extracts, which was in line with the expected size. The transfection efficiency of the lentivirus was assessed by fluorescence microscopy after transfection for 3 days, and GFP expression suggested successful transfection of activated HSCs by lentivirus (Fig. 1).
Histopathology of rat liver
After injection of pGC-FU-3FLAG-PPAR-γ vector, strong green fluorescence was detected in rat livers by confocal microscopy (Fig. 2). A similar green fluorescence was observed in livers from the blank vector group. This demonstrated that in vivo transfection of vector was successful. Hepatic fibrosis was in the S0 stage in the control group. Also, in the 2-week group, S1- or S2-stage fibrosis was evident in the hepatic fibrosis and blank vector groups. In the 8-week group, mostly S3- or S4-stage fibrosis was evident in the hepatic fibrosis and blank vector groups while in the PPAR-γ-transfected group the fibrosis was in stage S1 to S3 (Fig. 3A). Collagen deposition in the hepatic fibrosis and blank vector groups was enhanced compared to the control and PPAR-γtransfected groups by Masson staining (Fig. 3B).
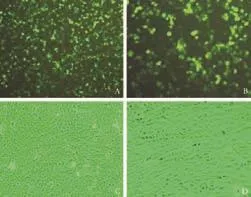
Fig. 1. A and B: Green fluorescence in HSCs from the PPAR-γoverexpression group 3 days after transfection. C and D: Activated HSCs cultured separately in 6-well plates for 14 days (A and C: original magnification ×100; B and D: original magnification ×200).

Fig. 2. Green fluorescence in liver from PPAR-γ-transfected group mainly located within and around fibrotic septa (original magnification ×100).
Expression of PPAR-γ,α-SMA and collageniprotein in activated HSCs and liver tissue
The expression of PPAR-γ protein in HSCs in the PPAR-γ-transfected group was higher than that in the lentiviral vector control or control groups (P<0.01). However, the expression of α-SMA and collageniprotein was down-regulated (P<0.01) (Fig. 4). The expression of PPAR-γ inhibited the activation of HSCs.
The in vivo study showed no difference between the expression of PPAR-γ protein in the PPAR-γtransfected group versus the blank vector group at the early stage of fibrosis, and it was enhanced compared to the hepatic fibrosis group (P<0.05). The difference was clear between the groups at the sustained fibrosis stage (P<0.01) (Fig. 5). There was no difference between the expression of collageniprotein in the PPAR-γtransfected group and the blank vector group or the hepatic fibrosis group at the early stage of fibrosis. The difference was significant between the groups at the sustained fibrosis stage (P vs. F, P<0.01; P vs. N, P<0.05) (Fig. 6). However in the early stage, the expression of α-SMA protein showed no difference between the PPAR-γ-transfected group and the blank vector group, but there was a difference between the PPAR-γ-transfected group and the hepatic fibrosis group (P<0.05). The difference was significant between the groups at the sustained fibrosis stage (P<0.01) (Fig. 7). These results suggested that the expression of PPAR-γ inhibited the development of hepatic fibrosis.
HSC proliferation using the MTT assay
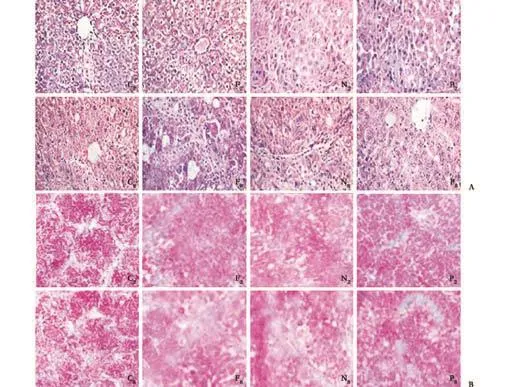
Fig. 3. A: HE staining of liver sections; B: Collagen deposition by Masson staining of liver sections (original magnification ×100).
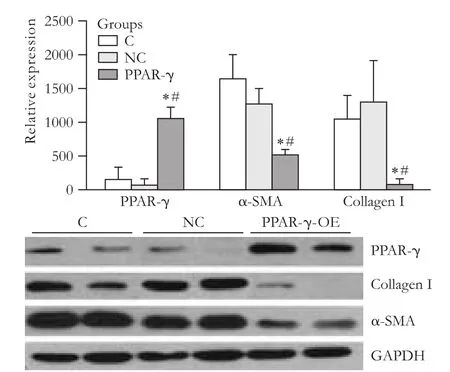
Fig. 4. Expression of PPAR-γ, α-SMA and typeicollagen protein in activated HSCs (*: P<0.01, vs. C group; #: P<0.01, vs NC group).
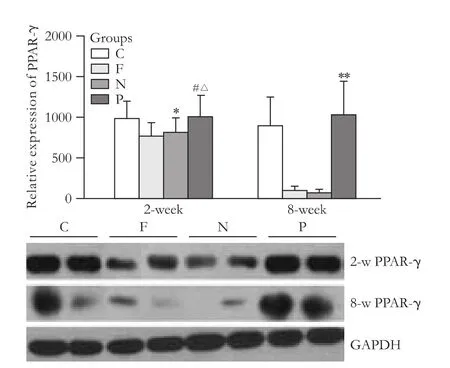
Fig. 5. PPAR-γ protein content in liver tissues of rats (*: P>0.05, vs. C and F groups; #: P>0.05, vs. C and N groups; △: P<0.05, vs. F group; **: P<0.01, vs. F and N groups).
To determine the role of pGC-FU-3FLAG-PPAR-γ transfection in the proliferation of HSCs, MTT was used to measure the proliferation of activated HSCs for 5 consecutive days after transfection. The results showed that 3 days after transfection, the proliferation ability of HSCs was reduced (P<0.01) (Fig. 8), suggesting that PPAR-γ inhibited the proliferation of activated HSCs.
Hydroxyproline content of liver tissue
At an early stage of fibrosis, the differences in the hydroxyproline content in the liver tissue of the hepatic fibrosis group, the blank vector group, and the PPAR-γ transfected group were not statistically significant. At the stage of sustained fibrosis, the differences in thehydroxyproline content were significant between the groups (P<0.01 and <0.05, respectively) (Fig. 9).
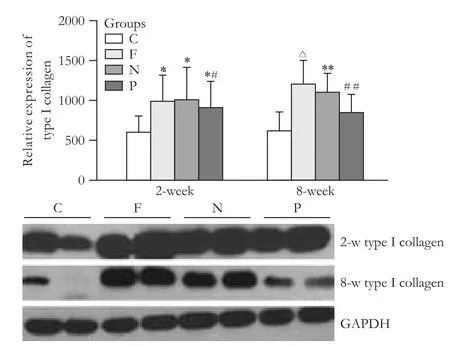
Fig. 6. Type i collagen protein content in liver tissues of rats (*: P<0.05, vs. C group; #: P>0.05, vs. F and N groups; (△: P<0.01, vs. P group; **: P<0.05, vs. P group; ##: P>0.05, vs. C group).
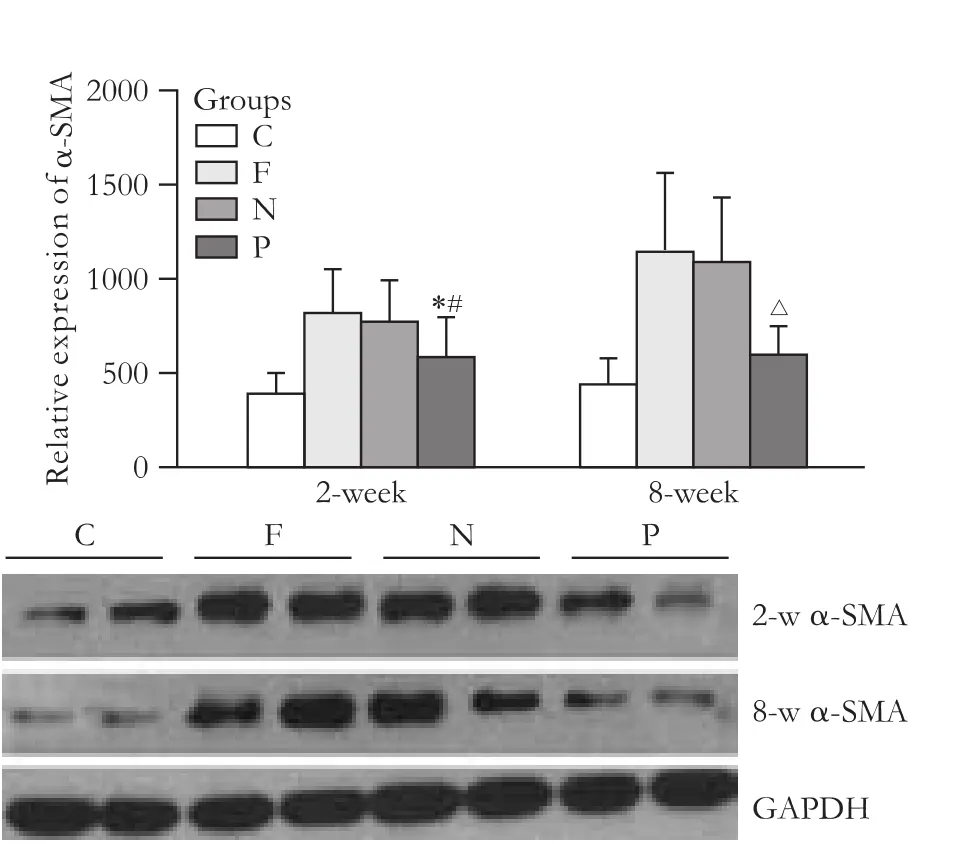
Fig. 7. α-SMA protein content in liver tissues of rats (*: P>0.05, vs. C and N groups; #: P<0.05, vs. F group; (△: P<0.01, vs. F and N groups).
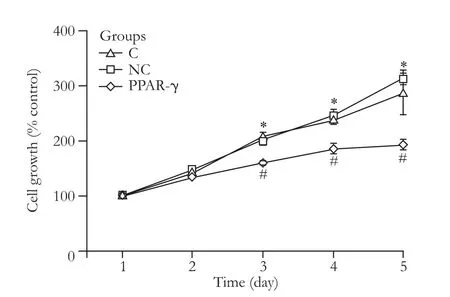
Fig. 8. Proliferation curve of activated HSCs (*: P>0.05, vs. NC group; #: P<0.01, vs. C and NC groups).
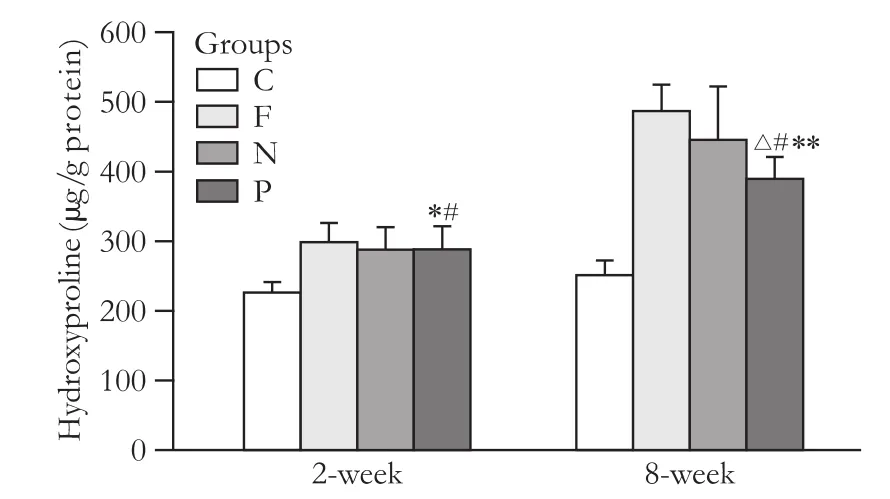
Fig. 9. Hydroxyproline content in liver tissue (μg/g protein) (*: P>0.05, vs. F and N groups; #: P<0.01, vs. C group; (△: P<0.05, vs. N group; **: P<0.01, vs. F group).
Discussion
HSCs, also known as extracellular matrix-producing cells, are considered the main source of extracelluar matrix during the process of hepatic fibrosis. After stimulation, HSCs lose the vitamin A lipid droplets and become myofibroblasts. The activated HSCs express α-SMA and may secrete collagen I, III, IV, laminin, fibronectin, hyaluronic acid and other extracellular matrix components. The increased expression of α-SMA is a marker of HSC activation and myofibroblast formation. After activation, the major secreted collagen is type I.
PPAR-γ is a nuclear transcription factor that plays an important role in fat formation, cell growth, cell differentiation and many other biological processes. PPAR-γ is expressed in quiescent HSCs, and its expression is significantly decreased after HSC activation.[13,14]Previous reports showed, during the process of HSC activation, a notable decline of the vitamin A content in these cells due to the decreased expression of fat-formation-related transcription factors such as PPAR-γ.[15]Bothin vivoandin vitrostudies have shown that PPAR-γ-specific agonists or ligands inhibit activation of HSCs and induce apoptosis of HSCs through a variety of signal pathways.[8,9,16]The induction of PPAR-γ expression can significantly improve fibrosis in the liver injury process.[17]Clinical studies have shown that the treatment of hepatic fibrosis with PPAR-γ agonists, such as rosiglitazone or pioglitazone can have desirable effects.[18]However, some reports showed that large doses of certain PPAR-γ ligands do not affect the PPAR-γ signaling pathway and also inhibit the proliferation of HSCs.[19]To accurately evaluate the role of PPAR-γ on inhibition of HSC activation and hepatic fibrosis, we constructed a PPAR-γ-expression plasmid mediated by lentivirus. Then, the plasmids were directly transfected into primary HSCs in vitro and a rat model of hepatic fibrosis in vivo to increase the expression of PPAR-γ. The expressions of PPAR-γ, α-SMA and typeicollagen in HSCs and liver tissue were assessed. The hydroxyproline content in liver tissue was measured, and a pathological examination was performed. The proliferation of activated HSCs was also determined. Thesein vivoandin vitrostudies were used to investigate the effects of PPAR-γ expression on fibrosis.
In our in vitro study the exogenous expression of PPAR-γ in HSCs reduced typeicollagen synthesis. Besides collagen synthesis, the inhibitory effects of PPAR-γ were seen on other key parameters of HSC activation, including protein expression of α-SMA. Similar effects were reported in a study showing that PPAR-γ overexpression in HSCs diminishes collagen synthesis.[9]In addition, this also suppresses the proliferation of HSCs. Therefore, the antifibrogenetic effect of PPAR-γ in vivo is due, at least in part, to a decreased accumulation of activated matrix-producing HSCs and collagen production, thereby blocking or reversing the progression of fibrosis.
Our study indicated that increased PPAR-γ expression in the liver did not display clear inhibitory effects on early fibrosis. There were no significant differences between the amount of hydroxyproline, the expression of typeicollagen, and the severity of fibrosis in the transfected group and the control group. The expression of α-SMA was significantly different compared to the hepatic fibrosis group. The increased PPAR-γ expression had a significant inhibitory effect in the sustained fibrosis group. Further, the content of hydroxyproline and the severity of fibrosis were significantly different among the groups. The expression of PPAR-γ, typeicollagen and α-SMA were also significantly different among the groups.
Some studies have shown that the PPAR-γ agonist pioglitazone is not applicable to treatment of the advanced CCl4-induced rat model of fibrosis (5 weeks after model generation) and is a poor choice for the treatment of fibrosis induced by bile duct ligation.[20]Other studies showed that virus vector-mediated PPAR-γ expression has a good inhibitory effect on the rat model of hepatic fibrosis induced by bile duct ligation. This difference is probably due to the different mechanisms of gene expression.[17]Pioglitazone stimulates the expression of PPAR-γ, which influences quiescent but not activated HSCs. A large number of HSCs are activated with the progression of fibrosis; therefore, the efficacy of pioglitazone decreases. The independent expression, however, is not influenced by cell activation status, so the efficacy does not decrease with the progression of the disease. In the 2-week treated group in our study, the short time induced a less severeif brosis, and the expression of PPAR-γ was not completely inhibited. Thus, there was no signi ficant difference in PPAR-γ expression between the PPAR-γ-transfected group and the blank-vector transfected group or the normal control group, which led to insigni ficant ef ficacy. In the 8-week group, because of the severe fibrosis in the control group, the inhibitory effects of transfection were good. Another study demonstrated the anti- fibrotic effects of PPAR-γ in methionine-choline-de ficient-induced fibrosing steatohepatitis for 8 weeks in mice.[21]
In summary, our study showed that PPAR-γ overexpression inhibited hepatic fibrosis. The PPAR-γ thiazole ketone agonists have adverse effects, so patients with liver diseases should take caution when using these drugs. Much investigation is needed for the clinical application of these drugs.
Funding:This work was supported by a grant from the Science and Technology Commission of Shanghai Municipality (No. 07JC14036).Ethical approval:The protocol was approved by the Ethics Committee of Renji Hospital, Shanghai Jaotong University School of Medicine, Shanghai, China.
Contributors:WZ, XJP and ZYC proposed the study. WZ wrote the first draft. CW analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. WZY and LM are the guarantors.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275:2247-2250.
2 Senoo H, Kojima N, Sato M. Vitamin A-storing cells (stellate cells). Vitam Horm 2007;75:131-159.
3 Bae MA, Rhee SD, Jung WH, Ahn JH, Song BJ, Cheon HG. Selective inhibition of activated stellate cells and protection from carbon tetrachloride-induced liver injury in rats by a new PPARgamma agonist KR62776. Arch Pharm Res 2010;33: 433-442.
4 Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF Jr, Motomura K, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem 2000;275:35715-35722.
5 Chen H, He YW, Liu WQ, Zhang JH. Rosiglitazone prevents murine hepatic fibrosis induced by Schistosoma japonicum. World J Gastroenterol 2008;14:2905-2011.
6 Nan YM, Fu N, Wu WJ, Liang BL, Wang RQ, Zhao SX, et al. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand J Gastroenterol 2009;44:358-365.
7 Sun K, Wang Q, Huang XH. PPAR gamma inhibits growth of rat hepatic stellate cells and TGF beta-induced connective tissue growth factor expression. Acta Pharmacol Sin 2006;27: 715-723.
8 Wang X, Huang G, Mei S, Qian J, Ji J, Zhang J. Overexpression of C/EBP-alpha induces apoptosis in cultured rat hepatic stellate cells depending on p53 and peroxisome proliferator-activated receptor-gamma. Biochem Biophys Res Commun 2009;380:286-291.
9 Hazra S, Xiong S, Wang J, Rippe RA, Krishna V, Chatterjee K, et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem 2004;279:11392-11401.
10 Knook DL, Seffelaar AM, de Leeuw AM. Fat-storing cells of the rat liver. Their isolation and purification. Exp Cell Res 1982;139:468-471.
11 Carter V, Cable HC, Underhill BA, Williams J, Hurd H. Isolation of Plasmodium berghei ookinetes in culture using Nycodenz density gradient columns and magnetic isolation. Malar J 2003;2:35.
12 Chevallier M, Guerret S, Chossegros P, Gerard F, Grimaud JA. A histological semiquantitative scoring system for evaluation of hepatic fibrosis in needle liver biopsy specimens: comparison with morphometric studies. Hepatology 1994;20:349-355.
13 Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002;122:1924-1940.
14 Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 2000;119:466-478.
15 Graham A. Curcumin adds spice to the debate: lipid metabolism in liver disease. Br J Pharmacol 2009;157:1352-1353.
16 Kang Q, Chen A. Curcumin suppresses expression of lowdensity lipoprotein (LDL) receptor, leading to the inhibition of LDL-induced activation of hepatic stellate cells. Br J Pharmacol 2009;157:1354-1367.
17 Yang L, Chan CC, Kwon OS, Liu S, McGhee J, Stimpson SA, et al. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 2006;291:G902-911.
18 Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297-2307.
19 Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, et al. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology 2000;31:101-108.
20 Leclercq IA, Sempoux C, Starkel P, Horsmans Y. Limited therapeutic efficacy of pioglitazone on progression of hepatic fibrosis in rats. Gut 2006;55:1020-1029.
21 Yu J, Zhang S, Chu ES, Go MY, Lau RH, Zhao J, et al. Peroxisome proliferator-activated receptors gamma reverses hepatic nutritional fibrosis in mice and suppresses activation of hepatic stellate cells in vitro. Int J Biochem Cell Biol 2010; 42:948-957.
July 10, 2010
Accepted after revision October 15, 2010
Author Affiliations: Department of General Surgery, Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200127, China (Wang Z, Xu JP, Zheng YC, Chen W, Sun YW, Wu ZY and Luo M)
Meng Luo, MD, Department of General Surgery, Renji Hospital, Shanghai Jiaotong University School of Medicine, Shanghai 200127, China (Tel: 86-21-58752345ext3633; Fax: 86-21-58395057; Email: luomeng@renji.com)
© 2011, Hepatobiliary Pancreat Dis Int. All rights reserved.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Hepatobiliary & Pancreatic Diseases International (HBPD INT)
- Primary hepatic carcinosarcoma
- Aberrant methylation frequency of TNFRSF10C promoter in pancreatic cancer cell lines
- Protective effects of glutamine preconditioning on ischemia-reperfusion injury in rats
- Effects of suppressing glucose transporter-1 by an antisense oligodeoxynucleotide on the growth of human hepatocellular carcinoma cells
- Hepatocyte differentiation of human fibroblasts from cirrhotic liver in vitro and in vivo