丁酸氢化可的松中有关物质检测方法的改进
2011-05-17张新勇霍立茹
张新勇,霍立茹
南京长澳医药科技有限公司,南京 210038
《中国药典》2005版中收录的关于丁酸氢化可的松有关物质的高效液相色谱法采用水-乙腈-冰醋酸(55︰45︰0.5)为流动相,检测波长为240 nm,理论塔板数不低于1500。我们通过对丁酸氢化可的松原料药的紫外光谱分析以及流动相和色谱柱的对比,对药典中关于该药的有关物质检测进行改进性研究,结果表明采用Diamonsil C18(250 mm×4.6 mm,5 μm 色谱柱,0.02 mol·L-1磷酸二氢钾溶液(用磷酸调节pH 值至3.5)-甲醇-四氢呋喃(20︰65︰15)为流动相,紫外检测波长为314 nm,不仅检出信息量大,而且杂质检出多,分离效果好,理论塔板数不低于2000,优于药典的规定。
1 仪器与药品
Yanaco微量熔点测定仪;紫外光谱分析仪;岛津UV-2550PC紫外可见分光光度计;高效液相色谱仪,Agilent 1100 HPLC Series及 H66MC型超声仪。
丁酸氢化可的松原料与对照品(自制)。
甲醇、乙腈为色谱纯;磷酸、磷酸二氢钾、磷酸二氢钠、四丁基溴化铵、辛烷磺酸钠、乙酸铵、四氢呋喃、醋酸钠和磷酸二氢铵为分析纯。
2 方 法
2.1 熔点的测定
取本品,研成细粉,依据《中国药典》2010年版二部ⅥC规定,测定其熔点。以195℃作为起始温度,以1.0℃·min-1的速率升温。最终得出20090401批样品的熔点为206.0~207.5℃,小于药典熔距(4℃)要求。
2.2 紫外吸收光谱
取本品,加乙醇制成每1 mL中约含8 μg的溶液,照分光光度法 (《中国药典》2010年版二部IV A)测定,结果见图1。本品在314 nm的波长处有最大吸收,在330 nm处有次大吸收,在260 nm的波长处有最小吸收。

图1 丁酸氢化可的松紫外扫描图
2.3 有关物质的检测
2.3.1 色谱条件的筛选色谱柱的选择 选择不同厂家、不同牌号色谱柱进行试验,以能有效地检出杂质,分离效果好,主成分保留时间适中的色谱柱,供检测本品有关物质使用。Diamonsil C18(250 mm×4.6 mm,5 μm)色谱柱分离效果好,主成分保留时间适中,比Kromasil色谱柱检出杂质多、检出主峰前的一个杂质。故选择此色谱柱。
2.3.2 流动相的选择本品根据 《中国药典》2010版,选择不同的流动相体系,并对流动相组成的比例以及盐溶液pH值进行适当调整。以分离效果较好,能有效地检出杂质,主成分保留时间适中的流动相作为本品的流动相。再从起始原料及中间体分离效果,峰形,主成分保留时间,可重复性,以及对色谱柱的保护等因素综合考虑,在几组流动相中,经比较最终选用0.02 mol·L-1磷酸二氢钾溶液(用磷酸调节pH 值至3.5)-甲醇-四氢呋喃(20:65:15)为流动相。
2.3.3 测定波长选择取本品适量,加流动相制成1 mg·mL-1的溶液,用高效液相色谱仪进行全波长扫描(190~400 nm)。将液相色谱仪二极管阵列检测器的波长,分别设定在330 nm和260 nm进行试验,并将结果进行对比分析,最终选择信息量比较大,能有效检出杂质的314 nm,作为本品的测定波长。
2.3.4 供试品浓度的确定取本品适量,加流动相分别制成每1mL中含0.0512、0.1024、0.2048mg的溶液,作为供试品溶液。进样量为20μL,记录色谱图。
试验结果显示,0.1024、0.2048 mg·mL-1的供试品溶液能有效地检出杂质。考虑到对色谱柱寿命的影响,故选定约0.1 mg·mL-1的溶液,作为供试品溶液的浓度。
2.3.5 色谱柱的理论板数及分离度在选定的色谱条件下,注入供试品溶液,记录色谱图,得出理论塔板数n=10665;分离度R=2.75,大于1.5。符合要求。见图2。
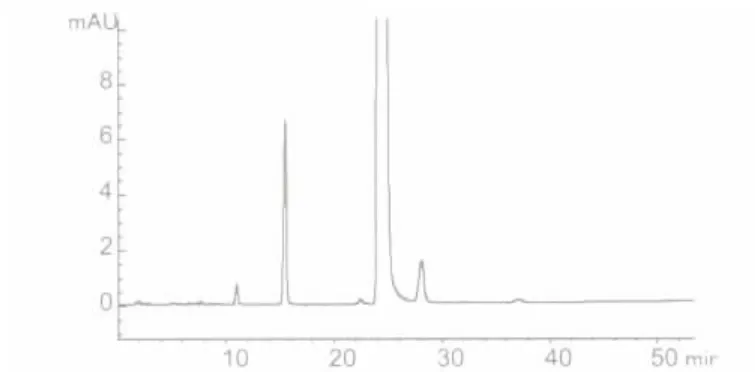
图2 改进后的丁酸氢化可的松液相色谱图
按照2010版《中国药典》采用水-乙腈-冰醋酸(55︰45︰0.5)为流动相,检测波长为240nm,进行检测,结果显示其检出信号少,有漏检或少检的可能。见图3。

2.3.6 检测限配制一定浓度的丁酸氢化可的松样品溶液,用流动相逐级稀释,进样,按3倍信噪比计算,测得丁酸氢化可的松最低检测限约为0.2ng·mL-1。
2.3.7 稳定性考察取本品适量,加流动相制成每1 mL中约含0.1 mg的溶液,作为供试品溶液,置室温自然条件下放置(避免强光照射),于不同时间,分别进样20 μL,记录色谱图,考察溶液的稳定性,结果表明,在4 h内测定液基本稳定。
2.3.8 精密度试验在选定的色谱条件下,取供试品溶液,连续进样6次,记录色谱图,量取峰面积,计算相对标准偏差RSD=1.12%,小于2%,符合规定。
2.3.9 破坏性试验取本品(批号20090401)进行强酸、强碱和氧化等破坏性试验,以峰面积归一化法,计算杂质含量。试验的目的是考察所设定的液相色谱条件,是否能有效地分离杂质,检出分解产物。
原溶液的配制 精密称取本品原料药25.5 mg,置25 mL量瓶中,甲醇-四氢呋喃(4︰1)溶解并稀释至刻度,摇匀;精密量取续滤液5 mL置50 mL量瓶中,加流动相稀释至刻度,摇匀,作为供试品溶液。进样,记录色谱图,作为0时对照。杂质总量为0.39%。
酸破坏 称取本品约25mg,置25mL量瓶中,加入5mol·L-1盐酸溶液适量,放置 2h,加甲醇-四氢呋喃(4︰1)溶解并稀释至刻度,摇匀,滤过;精密量取续滤液5mL置50mL量瓶中,加流动相稀释至刻度,摇匀,制成每1mL中含0.1mg的供试液,进样,记录色谱图,见图4A。试验结果表明,本品经酸破坏后测定,主峰与杂质峰及降解产物峰均有良好的分离。
碱破坏 称取本品约25mg,置25mL量瓶中,加入5mol·L-1氢氧化钠溶液适量,放置2h,加甲醇-四氢呋喃(4︰1)溶解并稀释至刻度,摇匀,滤过;精密量取续滤液5mL置50mL量瓶中,加流动相稀释至刻度,摇匀,制成每1mL中含0.1mg的供试液。进样,记录色谱图,见图4B。试验结果表明,本品经碱破坏后,主峰与杂质峰及降解产物峰均有良好的分离。
氧化破坏 称取本品约25 mg,置25 mL量瓶中,加30%H2O2溶液适量,振摇使破坏,加甲醇-四氢呋喃(4︰1)溶解并稀释至刻度,摇匀,滤过;精密量取续滤液5 mL置50 mL量瓶中,加流动相稀释至刻度,摇匀,制成每1 mL中含0.1 mg的供试液,见图4C。试验结果表明,本品经氧化破坏后测定,主峰与杂质峰及降解产物峰均有良好的分离。
上述试验结果,可以证明本品所建立的液相色谱条件与系统适用性良好,能有效地监控产品质量。
2.3.10 色谱图记录时间的考察依据本品原料药和强烈破坏性试验结果,色谱图记录至主成分峰保留时间的3倍即可,大约在30分钟各组分已被洗脱完毕。
2.3.11 样品杂质总量的测定色谱条件与系统实用性试验:用十八烷基硅烷键合硅胶为填充剂,0.02mol·L-1磷酸二氢钾溶液(用磷酸调节pH 值至3.5)-甲醇-四氢呋喃(20︰65︰15)为流动相,检测波长314 nm,理论板数以丁酸氢化可的松峰计算不低于2000。

测定法:取本品约25 mg,精密称定,置25 mL量瓶中,加甲醇-四氢呋喃(4︰1)溶解并稀释至刻度,摇匀,再加流动相稀释10倍作为供试品溶液;精密量取供试品溶液适量,用流动相稀释成每1 mL中含10 μg的溶液,作为对照溶液。精密量取供试品溶液与对照溶液各20 μL,分别注入液相色谱仪,记录色谱图至主成分色谱峰保留时间的3倍,供试品溶液色谱图中如有杂质峰,量取各杂质峰面积,计算有关物质的量。
[1] 国家药典委员会.中华人民共和国药典[S].北京:化学工业出版社,2005.
[2] 中国药品生物制品检定所.中国药品检验标准操作规程[M].北京:中国医药科技出版社,2005:128.
[3] 刘文英.药物分析[M].第6版.北京:人民卫生出版社,2007:83.
