2-甲酰基-4,6-二羟基苯甲酸甲酯及其衍生物的合成
2011-04-07马军营孙超伟白争辉
马军营,鲍 丰,孙超伟,白争辉,华 林
(河南科技大学化工与制药学院,河南洛阳471003)
0 前言
药理实验表明:许多间苯二酚类大环内酯(如Aigiacomycin A-E[1],Resor-cylide[2],Lasiodiplodin[3],Redicicol[4]等)和含有间苯二酚结构的天然产物(如Zearalenone[5],Pochnins A-F[6],Gustastatin[7]等)具有抗肿瘤、抗菌、抗疟疾及抗动物体内P388白血病等生理活性[8-10]。尤其是20世纪80年代,从海洋生物红树上的子囊菌类(Aigialus parvus)中提取的 Hypothemycin[1]和2002年 Isaka在 Aigialus parvus BCC5311的次生代谢物中分离的十四元环间苯二酚类大环内酯Aigiacomycin A-E[1],具有较强的抗疟疾活性,引起了有机化学工作者的研究兴趣,并进行了大量的合成工作。2-甲酰基-4,6-二羟基苯甲酸甲酯及其衍生物是合成这类间苯二酚类大环内酯的重要中间体[11]。为此,本文以乙酰乙酸甲酯为原料,拟采用直线型的合成方法,经芳环化、羟基酯化、氧化、水解、羟基甲醚化等反应,对2-甲酰基-4,6-二羟基苯甲酸甲酯及其衍生物进行了合成。
1 实验部分
1.1 仪器与试剂
熔点测定采用Kofler型熔点仪(未校正);核磁共振氢谱和核磁共振碳谱采用Bruker AM-300型核磁共振谱仪测定(TMS为内标);质谱数据用ZAB-HS型质谱仪测定。所用试剂均为市售分析纯或化学纯。柱层析硅胶(200~300目,试剂级)和薄层析硅胶(GF254,化学纯)为青岛海洋化工有限公司生产,绝对无水乙醚和四氢呋喃用钠砂加热回流干燥二苯甲酮检测变蓝,其他溶剂按标准方法处理。
1.2 合成思路与合成路线设计
根据2-甲酰基-4,6-二甲氧基苯甲酸甲酯及其衍生物的结构特点,首先以乙酰乙酸乙酯为原料,在强碱氢化钠和正丁基锂催化下,通过改进Vattas芳构化方法生成了2,4-二羟基-6-甲基苯甲酸甲酯(5);其在硫酸催化下与乙酸酐反应,生成2,4-乙酰氧基-6-甲基苯甲酸甲酯(4);化合物(4)在酸性条件下经三氧化铬氧化制得2,4-乙酰氧基-6-(二乙酰氧基甲基)苯甲酸甲酯(3);化合物(3)在甲醇中回流,得到目标产物2-甲酰基-4,6-二羟基苯甲酸甲酯(2)。为了拓展化合物(2)在间苯二酚类大环内酯合成中的适用范围,又将化合物(2)与碳酸钾和碘甲烷反应合成化合物(2)的二甲醚化产物——2-甲酰基-4,6-二甲氧基苯甲酸甲酯(1a),将化合物(3)在水合硫酸铁催化下回流得到化合物(2)的二乙酰氧基化产物——2-甲酰基-4,6-二甲氧基苯甲酸甲酯(1b)。基于此合成思路,2-甲酰基-4,6-二羟基苯甲酸甲酯及其衍生物的合成路线见图1。此合成方法具有原料价廉易得、操作简便、产率高等特点。
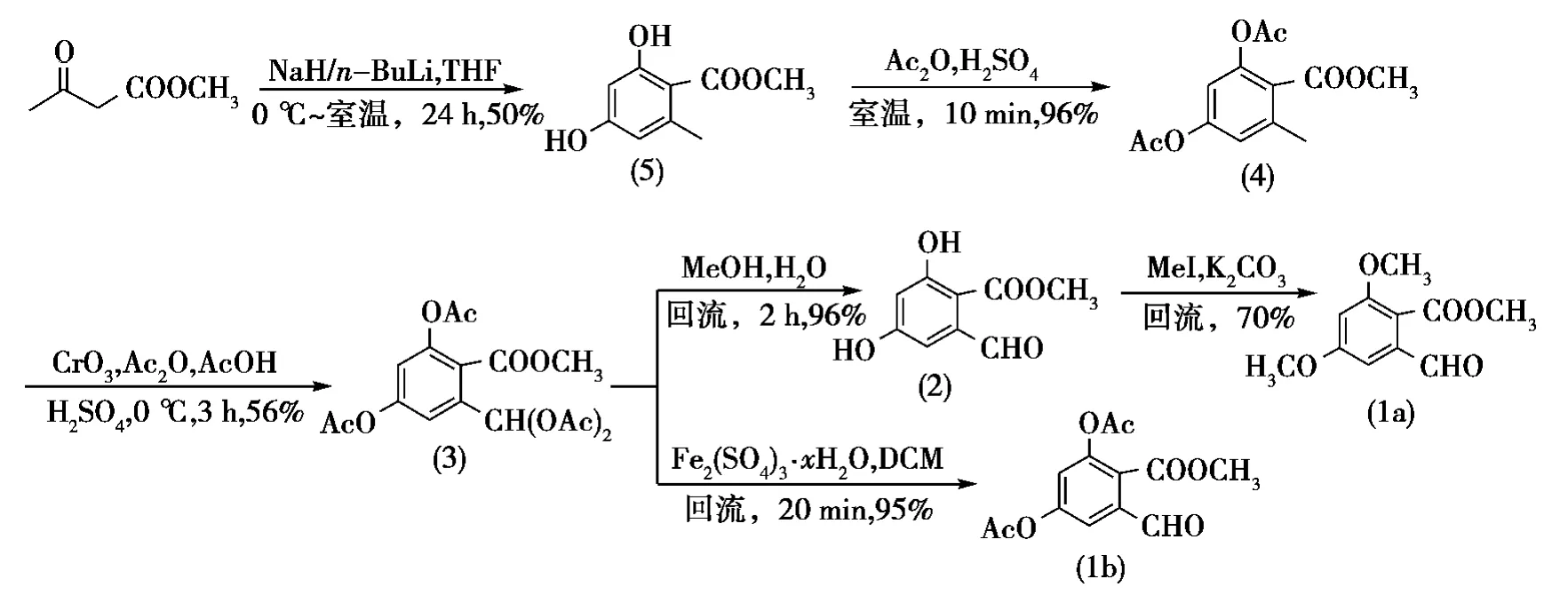
图1 2-甲酰基-4,6-二羟基苯甲酸甲酯及其类似物的合成路线
1.3 合成与结构表征
1.3.1 2,4-二羟基-6-甲基苯甲酸甲酯(5)的制备
在氩气保护下向250 mL三口烧瓶中分别加入质量分数为60%的NaH 6.60 g(165 mmol)和150 mL无水四氢呋喃,用冰水浴将温度控制在0℃,向反应体系中缓慢滴加17.41 g(150 mmol)乙酰乙酸甲酯。滴加完毕后继续在0℃下搅拌10 min,再缓慢滴加摩尔浓度为2.5 mol/L的n-Bu-Li溶液30 mL(75 mmol)后逐渐升至室温。反应24 h后用质量分数为10%的盐酸溶液淬灭反应,并使反应液呈酸性(pH=2)。加入少量固体氯化钠,用乙酸乙酯萃取(4×100 mL),有机相用饱和氯化钠溶液洗涤,无水Na2SO4干燥。过滤、减压蒸去溶剂和未反应的乙酰乙酸甲酯,残余液用乙酸乙酯重结晶得到2,4-二羟基-6-甲基苯甲酸甲酯(5)无色晶体6.83 g,产率为50%。熔点139~140℃(文献[12]为138~140℃)。1H NMR(300 MHz,CD3COCD3),δ:2.42(s,3H,CH3),3.89(s,3H,OCH3),6.23(s,1H,Ar-H),6.27(s,1H,Ar-H);13C NMR(75 MHz,CD3COCD3),δ:23.52,51.49,100.77,104.43,111.65,143.55,162.77,165.20,172.19;MS(质荷比):182(M+),150,122,94,77,69,39。
1.3.2 2,4-二乙酰氧基-6-甲基苯甲酸甲酯(4)的制备
向10 mL锥形瓶中分别加入3.66 g(20 mmol)2,4-二羟基-6-甲基苯甲酸甲酯(5)、6 mL(30.3 mmol)乙酸酐和两滴浓硫酸,快速搅拌使化合物(5)迅速溶解。10 min后待反应体系变澄清时,将反应液倾入盛有10.00 g碎冰的烧杯中,碎冰融化后有大量白色固体析出。抽滤,冰水洗涤,白色固体置于放有五氧化二磷的真空干燥器中干燥,得2,4-二乙酰氧基-6-甲基苯甲酸甲酯白色粉末状固体(4) 5.11 g,产率96%。熔点45~47℃。1H NMR(300 MHz,CDCl3),δ:2.26(s,3H,CH3CO-),2.28(s,3H,CH3CO-),2.40(s,3H,CH3),3.87(s,3H,CH3O-),6.80(d,1H,J=1.8 Hz,ArH),6.89(d,1H,J=1.8 Hz,ArH);13C NMR(75 MHz,CDCl3),δ:20.70,21.01,21.33,52.45,114.47,121.53,140.03,149.53,151.97,166.53,168.89,169.11;MS(质荷比):266(M+),235,224,193,182,150,122,94.77,43。
1.3.3 2,4-二乙酰氧基-6-(二乙酰氧基甲基)苯甲酸甲酯(3)的制备
向250 mL圆底烧瓶中分别加入5.32 g(20 mmol)化合物(4)、50 mL乙酸、34 mL乙酸酐和6 mL浓硫酸。在0℃搅拌下2 h内分批加入6.03 g(60mmol)CrO3,在0℃下反应3 h后,将反应物倾入盛碎冰的烧杯中,加入过量Na2S2O5,用乙酸乙酯萃取(3×100 mL),有机层分别用饱和Na2CO3溶液、水、饱和NaCl溶液洗涤,无水Na2SO4干燥。过滤,浓缩,残余液经柱层析(V(石油醚)∶V(乙酸乙酯)=10∶1)得2,4-二乙酰氧基-6-(二乙酰氧基甲基)苯甲酸甲酯白色固体(3)4.28 g,产率56%。熔点129~130℃。1H NMR(300 MHz,CDCl3),δ:2.11(s,6H,CH3CO-),2.27(s,3H,CH3CO-),2.31(s,3H, CH3CO-),3.88(s,3H,OCH3),7.06(s,1H,ArH),7.27(s,1H,ArH),7.96(s,1H,CH);13C NMR(75 MHz,CDCl3),δ:20.63,21.08,52.73,86.37,117.88,118.09,121.40,122.47,136.79,149.38,152.17,164.74,168.31,168.63;MS(质荷比):382(M+),340,298,280,266,237,196,165,136,108,77,69,43。
1.3.4 2,4-二羟基-6-甲酰基苯甲酸甲酯(2)的制备
向装有冷凝管的25 mL圆底烧瓶中分别加入0.38 g(1 mmol)化合物(3)、9 mL甲醇、2 mL H2O和一滴硫酸。加热回流2 h后,冷却至室温,加饱和NaHCO3溶液,调节pH=7,用乙醚萃取(3×20 mL)。有机层分别用水、饱和NaCl溶液洗涤,无水Na2SO4干燥。过滤,浓缩,残余液经柱层析(V(石油醚)∶V (乙酸乙酯)=2∶1)得2,4-二羟基-6-甲酰基苯甲酸甲酯(2)白色固体0.18 g,产率96%。熔点186~187℃。1H NMR(300 MHz,CD3CO-CDCl3),δ:3.81(s,3H,CH3O),4.07~4.16(br.,2H,-OH),6.22(s,1H,CHO),6.46(d,1H,J=1.8 Hz,ArH),6.53(s,1H,J=1.8 Hz,ArH);13C NMR(75 MHz,CD3COCDCl3),δ:52.87,104.22,104.77,108.13,137.73,161.90,162.45,169.08,169.37;MS(质荷比): 196(M+),165,151,137,123,108,81,77。
1.3.5 2,4-甲氧基-6-甲酰基苯甲酸甲酯(1a)的制备
向25mL圆底烧瓶中分别加入0.196 g(1mmol)化合物(2)、0.35 g(2.5mmol)K2CO3、10mL丙酮和0.14 mL(2.2 mmol)碘甲烷。加热回流4 h后,过滤,浓缩,用乙醚萃取(3×20 mL),有机相分别用水、饱和NaCl液洗涤,无水Na2SO4干燥。过滤,浓缩,残余液经柱层析(V(石油醚)∶V(乙酸乙酯)= 10∶1)得化合物(1a)粗产品。用丙酮重结晶得纯白色固体化合物(1a)0.15 g,产率70%。熔点114~ 115℃。1H NMR(300 MHz,CDCl3),δ:3.56(s,3H,OCH3),3.88(s,3H,OCH3),3.93(s,3H,COOCH3),6.11(s,1H,CHO),6.48(s,1H,ArH),6.57(s,1H,ArH)。13C NMR(75 MHz,CDCl3),δ:56.33,99.31,100.78,101.54,107.67,149.75,159.57,166.68,167.35;MS(质荷比):224(M+),223,209,193,179,165,149,106,91,77。
1.3.6 2,4-乙酰氧基-6-甲酰基苯甲酸甲酯(1b)的制备
向装有冷凝管的25 mL圆底烧瓶中分别加入0.196 g(0.5 mmol)化合物(3)、20 mL CH2Cl2和0.025 g Fe2(SO4)3x H2O。加热回流20 min后,反应混合物过滤除去催化剂,浓缩,残余液经柱层析(V(石油醚)∶V(乙酸乙酯)=10∶1)得到白色固体(1b)0.27 g,产率为95%。熔点120~125℃。1H NMR(CDCl3,300 MHz),δ:2.28(s,3H,CH3CO),2.29(s,3H,CH3CO),3.92(s,3H,OCH3),7.37(d,1H,J=2.1 Hz,ArH),7.51(s,1H,J=2.1 Hz,ArH),10.04(s,1H,CHO)。13C NMR(75 MHz,CDCl3),δ:20.53,20.88,52.93,120.47,121.92,124.23,136.50,149.40,152.28,164.57,168.09,168.34,188.65。MS(质荷比):280(M+),279,249,237,207,195,136,97,85,71,57,43。
2 结果与讨论
2.1 二乙酰基化合物(3)水解反应的处理
据文献[12]报道,2,4-二乙酰氧基-6-(二乙酰氧基甲基)苯甲酸甲酯(3)在乙醇/水体系中,以浓硫酸为催化剂水解可得到2,4-二羟基-6-甲酰基苯甲酸甲酯(2),但在得到的产物(2)的质谱图中发现M+(196)丰度很低,而质荷比(210)丰度很高。推测这是由于在乙醇-水中水解时,化合物(3)的甲酯与体系中的乙醇发生了酯交换反应转化为化合物(2)的乙酯异构体,导致质荷比(210)峰的出现。于是本文采用了甲醇代替乙醇进行水解反应,顺利得到了化合物(2)。
2.2 化合物(2)水解反应后处理方法的改进
据文献[12]报道,化合物(3)水解反应后处理的程序是先加饱和碳酸氢钠溶液中和至pH=2,高温减压除去甲醇和水得固体化合物,固体化合物再用乙醚研磨、过滤,用乙醚洗涤数次后合并有机相,经过浓缩后得到2,4-二羟基-6-甲酰基苯甲酸甲酯(2)。由于此操作过程易使化合物(2)在高温下氧化,收率降低(产率为65%)。为此,本文采用了液相萃取法,即用饱和碳酸氢钠溶液中和后,质量分数为10%的盐酸酸化至pH=3,再用乙醚萃取(4×20 mL),有机相分别用水和饱和氯化钠溶液洗涤,经过减压浓缩得到粗产品,收率达到96%,比文献[12]的方法收率提高了47.6%。
2.3 化合物(1b)的合成比化合物(1a)原子更经济
2,4-甲氧基-6-甲酰基苯甲酸甲酯(1a)是由化合物(3)经过水解、甲醚化得到,而2,4-乙酰氧基-6-甲酰基苯甲酸甲酯(1b)直接由化合物(3)水解得到,其也是重要的中间体,相对而言,化合物(1b)比化合物(1a)的合成原子更经济。
3 结论
以乙酰乙酸乙酯为原料,采用直线型合成方法,经芳环化、羟基酯化、氧化、水解等4步反应,以25.8%总收率得到了6-甲酰基-2,4-二羟基苯甲酸甲酯(2);为了拓展其应用范围,又将化合物(2)通过酚羟基保护、将化合物(3)在水合硫酸铁的催化下直接水解分别得到了适应不同反应条件的类似物(1a)和(1b);通过对Vlattas方法的改进,提高了6-甲酰基-2,4-二羟基苯甲酸甲酯的收率,并对所有合成的中间体和目标产物进行了核磁共振1H谱和13C谱及质谱的表征。
[1] ChoiH G,Son JB,Park D S.etal.An Efficientand Enantioselective Total Synthesis of Naturally Occurring L-783277[J].Tetrahedron Letters,2010,51:4942-4946.
[2] Napolitano C,Natoni A,Santocanale C,etal.Isosteric Replacementof the Z-enonewith Haloethyl Ketone and E-enone[J].Bioorganic&Medicinal Chemistry Letters,2011,21:1167-1170.
[3] Yang SX,Gao JM,Zhang Q.Toxic Polyketides Produced by Fusarium sp.,An Endophytic Fungus Isolated from Melia Azedarach[J].Bioorganic&Medicinal Chemistry Letters,2011,21:1887-1889
[4] Zeng J,Valiente J,Zhan J.Specific Inhibition of the Halogenase for Radicicol Biosynthesis by Bromide at the Transcriptional Level in Pochonia Chlamydosporia[J].Biotechnol Lett,2011,33:333-338.
[5] Zimmermann T J,Niesen FH,Pilka E S,etal.Discovery of a Potentand Selective Inhibitor for Human Carbonyl Reductase 1 From Propionate Scanning Applied to the Macrolide Zearalenone[J].Bioorganic&Medicinal Chemistry,2009,17:530-536.
[6] Barluenga S,Lopez P,Moulin E,et al.Modular Asymmetric Synthesis of Pochonin C[J].Angew Chem Int Ed,2004,43: 3467-3470.
[7] García F J,Debergh JR,Brabander J K.A Photochemical Entry to Depsides:Synthesis of Gustastatin[J].Org Letters,2005,7(4):685-8.
[8] Wang S,Xu Y Q,Maine E A,et al.Functional Characterization of the Biosynthesis of Radicicol,An Hsp90 Inhibitor Resorcylic Acid Lactone from Chaetomium Chiversii[J].Chemistry&Biology,2008,15:1328-1338
[9] Laurent D,Pietra F.AntiplasmodialMarine Natural Products in the Perspective of Current Chemotherapy and Prevention of Malaria[J].Marine Biotechnology,2006,8(5):433-447.
[10] Zeng J,Valiente J,Zhan J X.Specific Inhibition of the Halogenase for Radicicol Biosynthesis by Bromide at the Transcriptional Level in Pochonia Chlamydosporia[J].Biotechnol Lett,2011,33:333-338.
[11] James H W.The Chemistry of the Carbon-Transition Metal Double and Triple Bond:Annual Survey Covering the Year 2009[J].Coordination Chemistry Reviews,2011,255:3-10.
[12] Vlattas I,Harrison IT,Tokes L,et al.Cross,Synthesis of(±)-Zearalenone[J].JOrg Chem,1968,33:4176-4179.
