大肠杆菌中表达Cry1C基因及其重组表达产物的纯化及复性
2011-04-01曹思硕许文涛冉文君梁立兴贺晓云罗云波元延芳黄昆仑
曹思硕,许文涛,,冉文君,梁立兴,贺晓云,罗云波,元延芳,黄昆仑,,*
(1.中国农业大学食品科学与营养工程学院,北京 100083;2.农业部转基因生物食用安全监督检验测试中心(北京),北京 100083)
大肠杆菌中表达Cry1C基因及其重组表达产物的纯化及复性
曹思硕1,许文涛1,2,冉文君1,梁立兴1,贺晓云2,罗云波1,元延芳2,黄昆仑1,2,*
(1.中国农业大学食品科学与营养工程学院,北京 100083;2.农业部转基因生物食用安全监督检验测试中心(北京),北京 100083)
通过PCR方法将苏云金芽孢杆菌Cry1C基因从原始质粒中克隆出来。由于Cry1C基因携带了大量大肠杆菌稀有密码子,因此用PCR方法对其前86个密码子进行修改,以提高其在大肠杆菌BL21(DE3)中的表达量。Cry1C蛋白在大肠杆菌BL21(DE3)中以包涵体形式大量表达,将包涵体用8mol/L尿素溶解并用His TrapTMFF凝胶柱纯化。所得纯化蛋白在复性缓冲液中复性折叠,最终得到可溶并有生物活性(由三化螟活性实验验证)的蛋白。Cry1C蛋白纯度可达99.2%。
Cry1C蛋白;稀有密码子;包涵体;表达;纯化;复性折叠
苏云金芽孢杆菌是一种革兰氏阳性产孢细菌,该菌体能分泌一种特殊的抗虫晶体蛋白,被命名为δ-内毒素,是目前主要的生物农药之一[1]。苏云金芽孢杆菌的内毒素按氨基酸序列相似性被分为两类,Cry和Cyt δ-内毒素[2-3]。Cry毒素对多种有害昆虫幼虫具有毒性,如鳞翅目、双翅目、鞘翅目[4]、膜翅目、同翅目、直翅目、食毛目,线虫类、螨类和原生动物等[5-6]。此类毒素已经在商业化农业、森林管理和蚊虫控制中作为化学合成农药的替代品被广泛应用。Cry毒素还能被转入转基因植物中使其具有抗虫特性[1]。
自从Schnepf等[7]在1981年从HD21Dipel中克隆出第一个Cry基因以来,已有433种晶体蛋白基因被克隆。这些基因按照它们的氨基酸序列可以被分为49种,93个亚种,147类,328小类[2]。这些毒素其中Cry1A类毒素被广泛应用于转基因植物,致使有些幼虫对该种作物产生抗性。Cry1C毒素能有效抵抗鳞翅目害虫,可作为Cry1A毒素的替代物或与Cry1A毒素融合表达得到新的抗虫蛋白。
本实验通过PCR对Cry1C基因进行突变后将该基因连接到pET28a(+)载体上,在大肠杆菌的细胞质中进行表达。重组融合蛋白经过洗涤包涵体后用8mol/L尿素溶解,用His TrapTMFF凝胶柱纯化,之后在复性缓冲液中复性得到活性蛋白。此蛋白可被应用于Cry1C蛋白的安全性评价,为转Cry1C蛋白的水稻商业化生产提供参考依据。通过大量发酵生产的Cry1C蛋白还能作为生物农药以减少化学合成农药的应用。
1 材料与方法
1.1 菌株与质粒
含有Cry1C基因的质粒由华中农业大学林拥军老师惠赠;大肠杆菌DH5α、BL21(DE3)菌株及原核表达载体pET-28a(+)由本实验室保存。
1.2 酶与试剂
T4 DNA连接酶、pGEM-T载体 美国Promega公司;Taq DNA聚合酶、限制性内切酶 日本Takara公司;DNA Marker DL2000、DNA回收试剂盒 天根生化科技(北京)有限公司;低分子质量蛋白质Marker 北京全式金生物技术有限公司;凝胶柱 美国Amersham Biosciences公司;其他试剂均为国产分析纯。
1.3 Cry1C基因密码子优化
用修改后的上游引物5'-TGGAGGAGAACAA TCAGAACCAGTGTATCCCGTACAATTGTCTGA GCAATCCGGAAGAAGTTCTGCTGGATGGCG AACGCATCTC-3'和下游引物5'-TCATCACT TTTGTGCGCGTTCAAGGTCAGATTCTGC -3'从原质粒中扩增并修改Cry1C基因[8]。5'端的稀有密码子被替换结果如图1所示。用1%琼脂糖凝胶电泳检测并回收目的条带后通过T4 DNA 连接酶于4℃连接过夜至pGEM-T载体,然后转化至E. coli DH5α。转化株首先经过蓝白斑筛选,之后再用PCR鉴定及测序鉴定。被修改后的基因被命名为Cry1C-rcp基因。
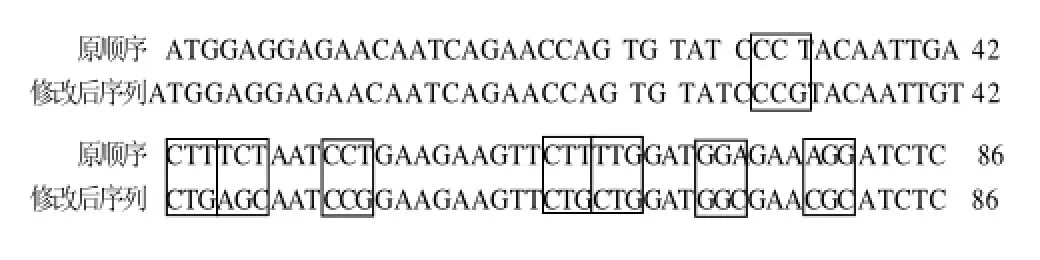
图1 Cry1C基因前86个碱基修改密码子Fig.1 Modified triplets in the first 86 nucleotides of Cry1C gene.
1.4 构建重组表达载体
通过带有酶切位点的上游引物 5'-GCGGATCCT CATCACTTTTGTGCGC -3'(下划线部分为BamH I 酶切位点)和下游引物5'-GGAATTCCATATG GAGG AGAACAATCAGAACC-3'(下划线部分为Nde I 酶切位点) PCR扩增Cry1C-rcp基因, PCR产物经BamH I 和Nde I酶切后插入pET-28a(+)的BamH I和Nde I位点。将此质粒转入大肠杆菌DH5α,提取转化株质粒经BamH I 和Nde I酶切鉴定及测序鉴定后,转入大肠杆菌BL21 (DE3)表达Cry1C蛋白。
1.5 表达重组蛋白
重组蛋白在含有100μg/mL卡那霉素的LB (1% 胰化蛋白胨、0.5%酵母提取物、1% NaCl)培养基中表达。将6mL的过夜培养物加入200mL的新鲜培养基中,37℃振荡培养。在OD600nm为0.6~1.0时考察不同IPTG浓度、温度和诱导时间对表达重组蛋白的影响。12000r/min离心10 min收集菌体,通过SDS-PAGE电泳分析得到最适的培养条件。
1.5.1 IPTG 诱导剂加入量的优化
当菌液OD600nm为0.6~1.0时向7个无菌小三角瓶中等量分装一定体积的菌液,然后依次向各瓶中加入IPTG至终浓度为0.01、0.05、0.1、0.2、0.5、1.0、2.0mmol/L。空载对照(即将pET-28a(+)质粒直接转入BL21(DE3)菌株中)加入IPTG 终浓度为1mmol/L。于37℃诱导表达4h,检测蛋白表达情况。
1.5.2 诱导温度的优化
当菌液OD600nm为0.6~1.0时等量分装于4个无菌三角瓶中,加入0.05mmol/L IPTG,分别于15、20、30、37℃条件下诱导表达4h 后收集菌体,检测蛋白表达情况。
1.5.3 诱导时间的优化
当菌液OD600nm为0.6~1.0时向6 个无菌小三角瓶中等量分装一定体积的菌液,然后加入IPTG终浓度为0.05mmol/L进行诱导表达。于37℃诱导6h,每隔1h 取样检测蛋白表达情况。
1.6 提取包涵体
取3g菌体加入20mL裂解液(50mmol/L Tris-HCl、10mmol/L EDTA、100mmol/L NaCl,pH8.0),加入溶菌酶至1mg/mL,37℃孵育30min,每隔10min搅动一次。冰浴超声波破碎,功率200~300W,每次10s,间隔10s,共30次。4℃、12000r/min离心20~30min沉淀包涵体,弃上清液。加入30mL洗涤液(50mmol/L Tris-HCl、10mmol/L EDTA、100mmol/L NaCl、0.5% Triton、10mmol/L β-巯基乙醇,pH8.0)超声波破碎,功率200~300W,每次10s,间隔10s,共10次。于4℃、12000r/min 离心20~30min沉淀包涵体,弃上清液;重复洗涤两次;加入20mL尿素洗涤液(100mmol/L Tris-HCl、1mol/L 尿素,pH8.0)再洗涤一次,得到纯度较高的包涵体蛋白。用无菌水洗涤包涵体两次,冻干,-80℃保存蛋白[9]。
1.7 纯化包涵体蛋白
将蛋白溶解于结合缓冲液 (20mmol/L NaH2PO4、40mmol/L 咪唑、0.5mol/L NaCl、8mol/L尿素,pH8.0)中。His TrapTMFF凝胶柱用10mL双蒸水洗涤后用10mL结合缓冲液平衡,之后使目的蛋白与凝胶柱结合。没有结合的蛋白用洗涤缓冲液(20mmol/L磷酸钠、100mmol/L咪唑、0.5mol/L NaCl、8mol/L尿素,pH8.0)除去。最后用洗脱缓冲液(20mmol/L磷酸钠、500mmol/L咪唑、0.5mol/L NaCl、8mol/L尿素,pH8.0) 洗脱,得到目的蛋白[9]。
1.8 复性重组蛋白
纯化后的Cry1C蛋白用透析缓冲液(pH 8.0,50mmol/L Tris-HCl,8、6、4、2、1、0.5、0mol/L尿素)4℃透析。每种尿素浓度需8h透析3次。重新折叠后的Cry1C蛋白在50mmol/L Tris-HCl (pH8.0) 缓冲液中于12000r/min离心15min以去除不溶蛋白。
1.9 SDS-PAGE定量蛋白含量
以牛血清白蛋白(BSA)为标准蛋白,用Bradford法检测样品中的蛋白含量[10]。将蛋白样品在上样缓冲液(50mmol/L Tris-HCl、8% 蔗糖、2% SDS、5%β-巯基乙醇、0.02% 溴酚兰)中煮沸5min后用SDS-PAGE电泳分离。电泳完毕后用考马斯亮蓝R-250染色,用脱色液(25%甲醇、10%乙酸)脱色至背景清晰为止。蛋白电泳图通过凝胶成像仪进行分析。
1.10 三化螟活性实验
三化螟的饲养方法参照文献[9],将包涵体蛋白、纯化后蛋白及复性后蛋白0~17.6μg/g与饲料混合。每个剂量做3个平行,每个试管接20头三化螟。
2 结果与分析
2.1 Cry1C基因密码子优化及克隆
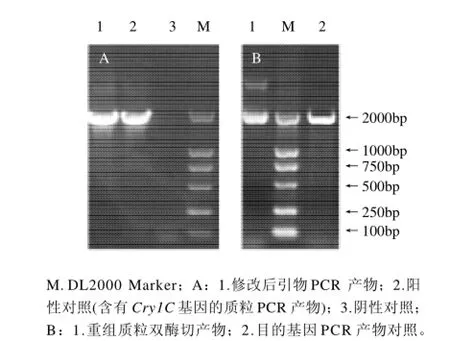
图2 PCR修改基因、重组质粒的酶切鉴定SDS-PAGE电泳图Fig.2 Agarose gel electrophoresis of PCR products amplified by modified upstream primers and agarose gel electrophoresis for the identification of recombinant plasmid digested with BamHI and Nde I
本实验通过设计PCR引物成功的将Cry1C基因中的大肠杆菌稀有密码子修改成大肠杆菌偏爱密码子,最终得到高效表达。修改后的引物PCR凝胶电泳结果如图2所示。将修改后的基因Cry1C-rcp通过引物加上BamH I和Nde I酶切位点,连入pET-28a(+)质粒载体,并转入大肠杆菌DH5α中,提取质粒经PCR酶切鉴定结果如图3所示。将鉴定为阳性的菌株送上海工生物工程技术服务有限公司测序,结果显示,Cry1C基因的全长1896碱基均无错配。将测序正确的质粒转入大肠杆菌BL21 (DE3)表达Cry1C蛋白。
2.2 表达条件优化
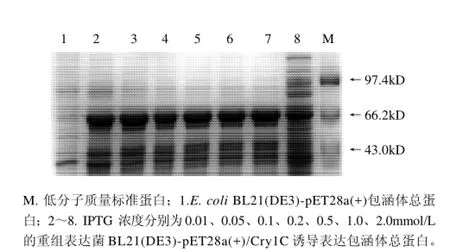
图3 IPTG浓度对Cry1C重组蛋白表达影响的SDS-PAGE分析Fig.3 Effect of IPTG concentration on recombinant Cry1C protein expression
从图3 可以看出,IPTG终浓度对目的蛋白表达量影响不大。在低浓度的IPTG诱导条件下(0.05mmol/L),目的蛋白已经有大量表达,随着IPTG浓度的增加,蛋白表达量增加不明显。
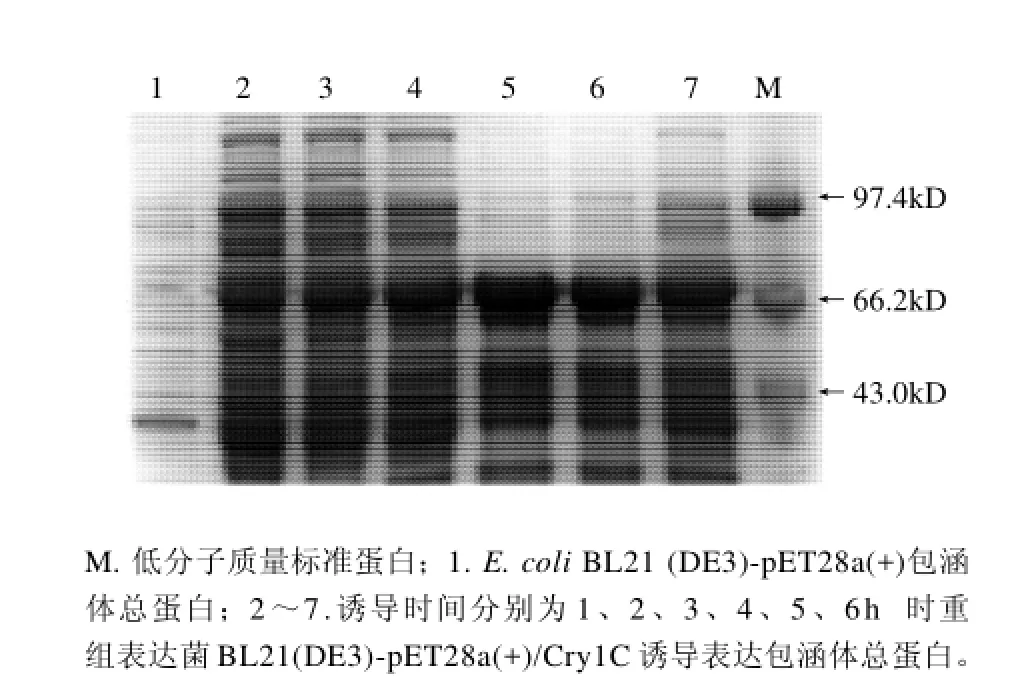
图4 诱导时间对Cry1C重组蛋白表达影响的SDS-PAGE分析Fig.4 Effect of induction time on recombinant Cry1C protein expression
从图4 可以看出,目的蛋白的诱导表达量随着诱导时间的延长而增加,当诱导时间达到4h后目的蛋白的表达量已经趋于稳定。有文献报道低温有利于蛋白的可溶性表达[11]。
本实验研究了在两个较低温度(15、20℃)及较高温度(30、37℃)条件下目的蛋白在菌体裂解液的上清液和包涵体中的表达情况。当诱导温度为15℃或20℃时,Cry1C目的蛋白有可溶形式表达;诱导温度为30℃或37℃时,目的蛋白几乎均以包涵体的形式存在。从蛋白表达的总量上看,37℃时得到的目的蛋白最多的(图5)。
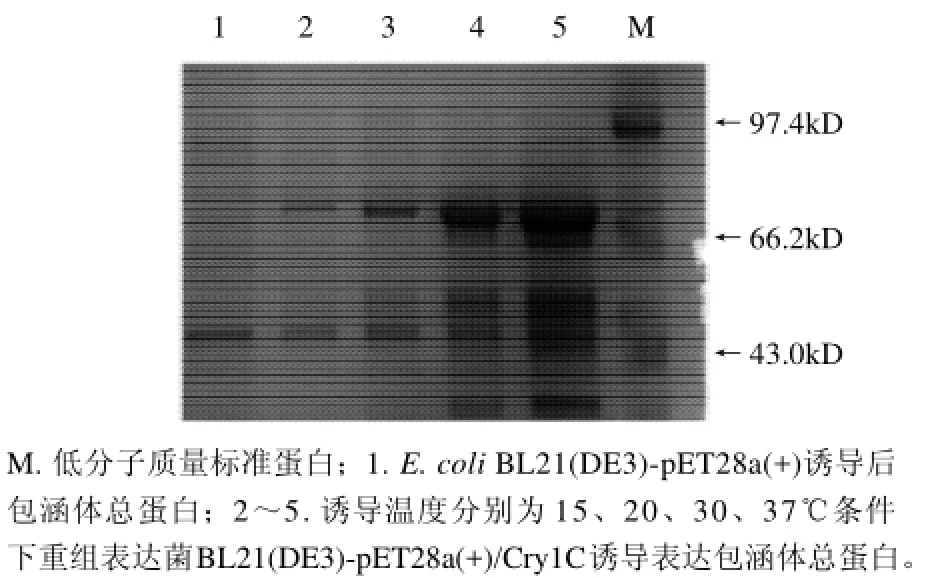
图5 诱导温度对Cry1C重组蛋白表达影响的SDS-PAGE分析Fig.5 Effect of induction temperature on recombinant Cry1C protein
2.3 复性及纯化
将IPTG终浓度为0.05mmol/L、37℃诱导4h的菌体用超声波破碎后,于12000r/min离心20min,取包涵体沉淀进行SDS-PAGE电泳,结果如图6所示。经过软件分析得出Cry1C蛋白条带的灰度占包涵体总蛋白灰度的52.9%,经过3次缓冲液及双蒸水的洗涤后目的蛋白的纯度可达86.7%,His TrapTMFF凝胶柱纯化后的蛋白纯度可达95.6%,透析后蛋白纯度没有太大变化,为95.8%。
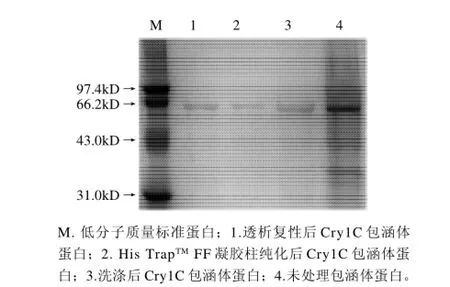
图6 Cry1C包涵体蛋白纯化复性SDS-PAGE电泳图Fig.6 SDS-PAGE of purified and refolded Cry1C protein
2.4 蛋白活性鉴定
由表1可见,复性后的蛋白表现出较高的杀虫活性,按1.10节的方法测定其杀虫活力,其LD50为4.99μg/g。包涵体蛋白和纯化后蛋白的LD50分别为8.36μg/g和 6.99μg/g。这表明本实验的蛋白纯化和复性方法能有效地纯化Cry1C蛋白和恢复其活性。

表1 Cry1C蛋白三化螟杀虫活性Table 1 Killing activity of different forms of Cry1C toxin against yellowstemborer
3 讨 论
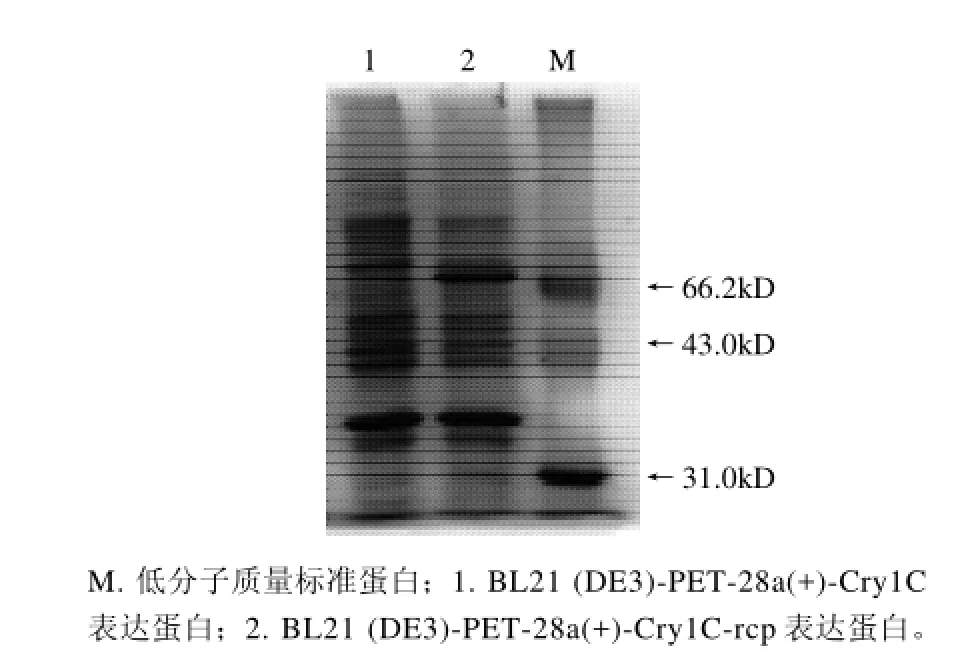
图7 稀有密码子修改前后Cry1C蛋白表达电泳图Fig.7 SDS-PAGE of Cry1C protein encoded by unmodified and modified rare condons
基因工程技术中影响外源基因在工程菌中表达效率的因素主要包括控制外源基因转录的启动子强弱、核糖体结合位点与起始密码子ATG之间的距离、外源基因的密码子在宿主细胞中的使用频率等。其中遗传密码子的偏性被认为是最为重要的影响因素。大肠杆菌和真核生物相比,其氨基酸密码子的使用频率不同,存在着一个明显的倾向性,并且细胞内对应的 tRNA 量与密码子的使用频率成正比。常用密码子(majar codon)是指那些在高表达基因中出现的密码子,而稀有密码子(rare codon)是指那些低水平表达的基因中出现的密码子,当E. coli高表达外源蛋白时,如果信息密码子的分配情况与E. coli相似,则细胞能正常进行 tRNA的乙酰化和蛋白翻译,减少翻译错误出现的频率,反之如果信使RNA带有稀有密码子,则蛋白质的翻译会出现障碍,影响基因的表达水平,在E. coli的mRNA中,AGG/ AGA出现的频率较低,分别近似为 0.14%和 0.21%,它们对外源蛋白表达的影响是近年来关于稀有密码子研究的热点之一[12]。另外有文献报道稀有密码子的位置要比它的频率更能影响蛋白表达。在5'端25个碱基内的稀有密码子会影响蛋白表达[13-15]。本实验正是通过修改前86个碱基中的稀有密码子以实现在大肠杆菌中的稳定、高效表达。本实验也尝试了常规表达方法(图7),但是未见外源蛋白表达。另外,本实验还应用了BL21 (DE3)-CodonPlus进行表达,但是表达情况并无明显改善。
实质等同性原则注重于转基因植物与传统对比物差异的评价,转基因植物外源基因表达蛋白的安全性评价是现阶段转基因植物商品化的主要技术问题之一。目前的分离技术尚不能从转基因植物中分离纯化出足够用于安全性评价的表达蛋白质产物,因此,可以用外源表达系统产生目的蛋白质。凡是以微生物表达的蛋白代替转基因植物中的目的蛋白,需要证实其与植物中的蛋白具有实质等同性方可使用。本实验在生物活性方面证实了两种蛋白的等同性。大肠杆菌表达的蛋白与植物蛋白在SDS-PAGE凝胶上大致处于同一位置,分子质量相近,生物活性实验结果证实大肠杆菌BL21(DE3)表达系统产生的目的蛋白与转基因植物中的目的蛋白基本是等同的,可以用于后续的食用安全性评价。
本实验实现了CryC蛋白的大肠杆菌表达、纯化及复性,并证实了该蛋白与水稻中该蛋白的实质等同性。CryC蛋白不仅能够用来代替水稻中的该蛋白来进行转基因作物的食用安全性评价,还能作为抗原制作抗体,实现CryC蛋白快速检测。另外通过大规模发酵还能生产生物农药,从而降低化学农药的使用,减少环境污染。
[1] SCHNEPF E, CRICKMORE N, van RIE J, et al. Bacillus thuringiensis and its pesticidal crystal proteins[J]. Microbiol Mol Biol R, 1998, 62: 775-806.
[2] CRICKMORE N, ZEIGLER D R, FEITELSON J, et al. Revision of the nomenclature for the Bacillus thuringiensis pesticidal crystal proteins [J]. Microbiol Mol Biol R,1998, 62: 807-813.
[3] HOFTE H, WHITELEY H R. Insecticidal crystal proteins of Bacillus thuringiensis[J]. Microbiol Mol Biol R,1989, 53: 242-255.
[4] BEEGLE C C, YAMAMOTO T. History of Bacillus thuringiensis berliner research and development[J]. Can Entomol, 1992, 124: 587-616.
[5] FEITELSON J S, PAYNE J, KIM L. Bacillus thuringiensis: insects and beyond[J]. Bio Technology, 1992, 10: 271-275.
[6] FEITELSON J S. The Bacillus thuringiensis family tree[M]//KIM L. Advanced engineered pesticides. New York: Marcel Dekker Inc., 1993: 63-71.
[7] SCHNEPF E, WHITELEY H R. Cloning and expression of the Bacillus thuringiensis crystal protein gene in E. coli[J]. Proc Natl Acad Sci USA, 1981, 78: 2893-2897.
[8] XU Wentao, HUANG Kunlun, WANG Ying. A cotton-specific gene, stearoyl-ACP desaturase, used as a reference for qualitative and real-time quantitative polymerase chain reaction detection of genetically modified organisms[J]. J Sci Food Agr, 2006, 86: 1103-1109.
[9] CAO Sishuo, XU Wentao, LUO Yunbo, et al. Expression, purification and refolding of recombinant Cry1Ab/Ac obtained in Escherichia coli as inclusion bodies[J]. Sci Food Agric, 2009, 89: 796-801.
[10] BRADFORD M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding[J]. Anal Biochem, 1976, 72: 249-254.
[11] 巩艳, 叶治家, 甘立霞, 等. sIL-16在大肠杆菌中的表达及活性分析[J]. 生命科学研究, 2002, 6(4): 318-321; 355.
[12] MAKRIDES S C. Strategies for achieving high-level expression of genes in E. coli[J]. Microbiol Mol Biol R, 1996, 60: 512-538.
[13] CHEN G F T, INOUYE M. Suppression of the negative effect of minor arginine codons on gene expression; preferential usage of minor codons within the first 25 codons of the E.coli genes[J]. Nucleic Acids Res, 1990, 18: 1465-1473.
[14] GAO Wenwu, TYAGI S, RUSSELL F, et al. Messenger RNA release from ribosomes during 5'-translational blockage by consecutive lowusage arginine but not leucine codons in Escherichia coli[J]. Mol Microbiol, 1997, 25: 707-716.
[15] GOLDMAN E, ROSENBERG A H, ZUBAY G, et al. Consecutive lowusage leucine codons block translation only when near the 50 end of a message in E. coli[J]. J Mol Biol, 1995, 245: 467-473.
Expression in E. coli, Purification and Refolding of Recombinant Cry1C Gene from Bacillus thuringiensis
CAO Si-shuo1,XU Wen-tao1,2,RAN Wen-jun1,LIANG Li-xing1,HE Xiao-yun2,LUO Yun-bo1,YUAN Yan-fang2,HUANG Kun-lun1,2,*
(1. College of Food Science and Nutrition Engineering, China Agricultural University, Beijing 100083, China;2. Supervision, Inspection and Testing Center of Genetically Modified Food Safety, Ministry of Agriculture, Beijing 100083, China)
The Cry1C gene from Bacillus thuringiensis (Bt) was cloned by polymerase chain reaction (PCR). Due to a large number of E. coli low-usage codons in the gene, the first 86 bases were optimized by PCR to improve the expression in E. coli. Cry1C protein was highly expressed in E. coli BL21 as inclusion bodies, which can be dissolved in 8 mol/L urea and purified by His TrapTMFF crude column under denaturing conditions. The purified Cry1C protein was dialyzed against the refolding buffer to obtain a soluble and biologically active protein. Finally, the protein was determined to be 99.2% purity.
Cry1C protein;rare codon;inclusion body;expression;purification;refolding
Q344.13
A
1002-6630(2011)07-0211-05
2010-06-02
农业部转基因生物重大专项(2008ZX08011-005;2009ZX08011-001B)
曹思硕(1984—),女,博士研究生,研究方向为食品安全。E-mail:cauxwt@yahoo.cn
*通信作者:黄昆仑(1968—),男,教授,博士,研究方向为食品安全。E-mail:hkl009@163.com
