长牡蛎(Crassostrea gigas)EST串联重复序列的组成和分布
2011-03-14张琳琳张国范
张琳琳,李 莉,张国范
(1.中国科学院 海洋研究所,山东 青岛 266071;2.中国科学院 研究生院,北京 100049)
长牡蛎(Crassostrea gigas)也称太平洋牡蛎,具有体型大、生长快、产量高、适应性强等优点,在我国北部沿海大面积养殖,是我国双壳贝类养殖中规模大、产量高的养殖品种之一。长牡蛎作为冠轮动物超门的模式种,在大片段文库和遗传图谱的构建,表达谱差异分析,杂种优势探讨等方面进行了较详细的研究[1-4],但基于大规模数据的重复序列方面的研究相对较少[5-6]。本文主要对长牡蛎EST进行串联重复序列结构类型,分布,丰度等的比较分析。
串联重复序列是指 1~200个碱基左右的核心重复单位,以头尾相串联的方式重复多次所组成的重复序列。它们在基因组中有着基因表达调节,群体遗传多样性分析等重要作用,与多种疾病相关[7]。 而简单序列重复,即微卫星(Simple sequence repeat,SSR),更是广泛地应用于遗传连锁图谱构建[8-9]和物种基因组结构的分析[10]。虽然长牡蛎大规模系统的基因组测序工作还没有完成,但NCBI上公布了大量的长牡蛎EST(Expressed sequence tags,表达序列标签)数据。所谓EST是指通过对cDNA文库随机挑取克隆进行大规模测序所获得的cDNA的5’或3’端序列,长度一般为150~500bp。研究表明长牡蛎EST中存在大量重复序列,可用于SSR标记的开发[6],这为从EST中寻找并分析串联重复序列提供了依据。通过物种间和物种内串联重复序列的比较,研究转录本的结构特征,分析其串联重复序列特别是 SSR的分布特征和可能的功能,将有助于了解基因组的起源和进化,同时更好地发挥这些序列在串联重复序列标记方面的应用。
截至2009年11月1日,在NCBI数据库中已登录了57 139条长牡蛎ESTs,但未有对上述57 139条EST全面的串联重复序列的报道。本研究旨在对现有长牡蛎EST中的串联重复序列信息进行结构类型,分布和丰度比较分析,以明确长牡蛎串联重复序列的发生频率和特点。同时分析了SSR在全长cDNA中的分布特点,以探讨长牡蛎转录本的结构和进化压力。本研究有助于促进串联重复序列特别是 SSR标记在基因组结构进化和长牡蛎遗传育种中的应用。
1 材料与方法
1.1 长牡蛎EST序列的下载和预处理
从NCBI库中下载57 139条长牡蛎ESTs(2009-11-01),过滤长度小于 100 bp的序列并与 UniVec(http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html)比对去除载体序列,在去掉3’末端的PolyA后,得到56 968条序列。利用 Sequence Assembly Program,CAP3[11]对上述序列进行初步聚类,采用的参数为重叠长度阈值N>30,重叠的一致性百分比N>90。
1.2 长牡蛎EST串联重复序列的分析
利用Tandem Repeat Finder (TRF)[12]对预处理的EST进行串联重复序列寻找,比对参数(匹配,不匹配,插入缺失)为 2,7,7,最小比对分值 30,重复单位最大长度500。过滤掉重复序列长度不足15bp的重复序列。如果同一位置出现的不同重复序列预报,本研究取重复序列长度最大的类型。长牡蛎的HindIII卫星序列的多序列比对采用 DNAMAN5.2.2(Lynnon Biosoft Company)。
1.3 长牡蛎 EST 5’UTR,3’UTR 和编码区CDS SSR分析
从NCBI库中下载644条长牡蛎蛋白质序列对应的EST序列,手工筛选出含有编码区全长和5’UTR,3’UTR的序列,共 80条。分别使用 TRF分析其5’UTR,3’UTR和CDS中SSR的分布情况。
2 结果
2.1 长牡蛎EST卫星重复序列的分析
在处理后的长牡蛎EST中共有10 997条串联重复序列(397 019 bp),其中小卫星重复序列(7~436 bp)有8 392条,共335 207 bp,占分析EST序列的1.58%(图1a,b,c)。重复序列单元总数目和重复类型间有一定规律性。重复序列单元总数目较多集中到7~12 bp,其中9 bp重复单元数目最多,为3 067个重复单元,其次是8 bp,10 bp,11 bp,12 bp,7 bp。从13 bp重复类型开始,重复单元数目降至1 000以下。随着重复单元长度的不断增加,重复单元数目大致上不断减少。在24~50 bp重复之间,重复单元数目波动相对较大。重复单元长度大于55 bp的区域中,在63 bp时出现一个峰,重复单元数目为 32.4,其他的重复单元类型相应的重复单元数目均小于25 bp。重复单元长度大于300 bp的只有3个重复类型,相应的重复单元总数目为 6.6。另一方面,串联重复序列平均拷贝数与重复类型并没有表现出线性关系,而是呈现不规律性的波动(图1 d)。
在长串联重复序列的分析中(本文中指串联重复序列的长度大于 100bp的重复类型),162~167bp 重复单元呈现一个明显的峰(图1 c)。将此部分序列提出,分析发现与长牡蛎的HindIII satellite DNA具有保守性(图2)。
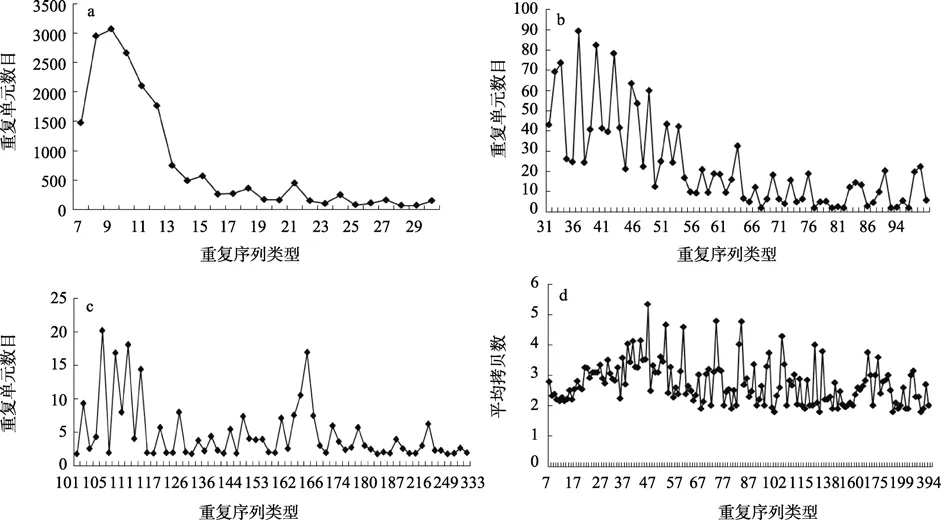
图1 不同串联重复序列类型在长牡蛎中的拷贝数特征Fig.1 The copy number of tandem repeats in the pacific oyster ESTs
2.2 长牡蛎EST简单串联重复序列的分析
长牡蛎 EST中含有丰富的 SSR,共 2 602个,61 744 bp,占分析序列总碱基的0.29%(表1)。重复序列数目表现为六碱基重复序列>单碱基>二碱基>三碱基>五碱基>四碱基,分别为851,805,307,258,240和141。重复序列长度、简单重复序列类型与拷贝数的研究过程中,发现重复序列单元长度与平均拷贝数成反比。另一方面,相同重复单元长度不同重复类型的重复序列数目、重复序列长度和平均拷贝数有很大的差别。每种重复单元类型代表其互补或顺序不同的所有重复单元,如 ATC代表ATG/TGA/GAT/CAT/ATC/TCA 6种重复类型。由于四碱基、五碱基、六碱基重复序列的重复类型较多,我们用 AT的百分比代替分析 SSR的分布特征和结构[9]。研究发现,A串联重复远远大于T。对于G串联重复最大拷贝数为 974,是因为 NCBI号为 FP000596的序列低质量测序,在除去此序列的影响后,G串联重复序列的最大拷贝数为 26。不同的重复序列重复类型重复序列的拷贝数目不同,如二碱基重复中,AG的重复序列数目远远大于AT、AC和GC。相同重复单元长度不同重复类型的平均拷贝数也有很大差别,并且与该重复类型的重复序列数目无关,如 ATC重复类型的重复序列数目约为ACT的30倍,但ACT重复类型的平均拷贝数大于ATC重复类型。
EST-SSR在标记应用时,多是以PCR为基础的,对SSR两侧的侧翼序列有一定长度的要求。因此,本研究统计了简单重复序列两侧的侧翼序列不低于30bp的微卫星位点,统计表明长牡蛎有1 954个简单重复序列位点符合要求,这些位点是微卫星标记开发的候选。

图2 长牡蛎HindIII 卫星序列的多序列比对Fig.2 Alignment of multiple HindIII satellites in Pacific oyster
2.3 长牡蛎cDNA 5’UTR,3’UTR和编码区CDS SSR分析
对挑选的含有5’UTR,3’UTR以及完整的编码区的80条长牡蛎序列分析发现UTR区域SSR长度所占的比例(0.005和 0.0026)远远大于 CDS区域 SSR所占的比例(0.0011)(表2)。因为5’UTR序列总长度相对较少,SSR 重复单元数目的关系为:5’UTR <CDS < 3’UTR,分别为 19,32.9 和 64.3。此外,cDNA的位置对简单串联重复序列的重复类型具有选择性。5’UTR区域只含有单碱基重复单元,CDS区域只含有三碱基倍数重复单元(三碱基/六碱基),3’UTR所含的重复单元类型较为丰富,含有单碱基,二碱基和五碱基重复单元。
3 讨论
3.1 长牡蛎EST中串联重复序列类型丰富,162~167bp类型含量高
从NCBI上下载的长牡蛎的EST序列中含有丰富的串联重复序列类型。覆盖从 1~436bp重复类型的 152种。对长牡蛎 100bp的重复类型中 162~167范围的峰值的分析表明,14个重复序列中有5个与长牡蛎的HindIII卫星序列具有高的相似度。南极贝(Adamussium colbecki)中曾报道了一个170bp重复单元的卫星序列,占基因组序列的 0.2%[13]。该卫星序列之后又被证明在牡蛎中具有中间的保守性,与哺乳动物的CENP-B box具有保守性,并被用来做牡蛎物种分类的标记[14]。
3.2 长牡蛎SSR分布广泛,1 594个候选位点
在简单重复序列中,从单碱基重复到六碱基重复均覆盖大多数重复序列类型。不同的简单重复序列类型的拷贝数目有很大差异。在二碱基重复中,AG的重复序列数目高达221,AT和AC均不超过50,GC最少为0,这与前人的报道相一致[8,15-16]。在三碱基重复序列中,ATC重复序列数目最多为73次,其次为AAC,AAT,AAG,AGG,其他的类型重复次数均小于15次,这与之前在栉孔扇贝中的报道类似[15]。在四、五、六碱基重复序列中,我们发现第二高AT百分比的重复序列类型拥有更高的重复序列数目,这与家蚕中的报道相一致[9]。从引物设计的角度考虑,
有1 594个位点为微卫星标记开发的候选位点,该结果为进一步开发长牡蛎EST-SSR标记奠定了基础。
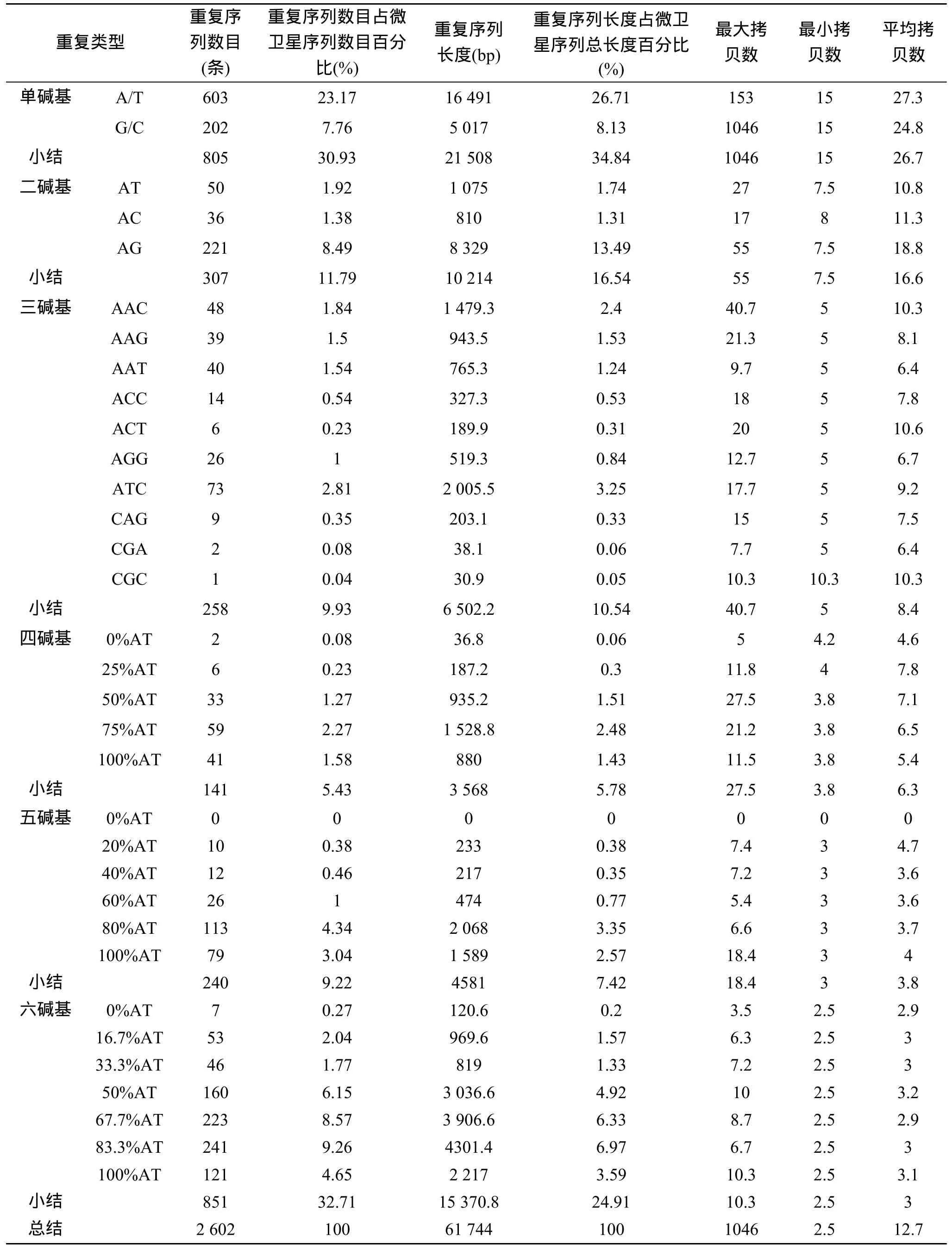
表1 长牡蛎EST微卫星重复序列的数目、长度和拷贝数特征Tab.1 The number,length,and copy number of SSR in the Pacific Oyster EST

表2 长牡蛎EST简单重复序列5’UTR,3’UTR和CDS特征Tab.2 The distributions of 5’UTR,3’UTR and CDS of SSR in the Pacific Oyster EST
3.3 长牡蛎EST的CDS区承受更大的选择压力
CDS区域简单串联重复序列相对较少,这与编码区受到的选择压力大于UTR区域有关,而编码区的重复序列类型为三碱基和六碱基,这两种碱基类型均为编码氨基酸的密码子数目3的倍数,这更说明了非3倍数的简单重复序列对编码区具有破坏作用,而自然选择将这部分破坏的简单重复序列淘汰了,这与水稻中的报道相一致[17]。在本研究中,编码区三碱基重复序列的类型为ACA,GAA和GAT重复,推测该三种重复类型可能与串联重复数目具有一定联系,其进一步研究可能需要使用更多的全长cDNA才能得出更明确的结论。
[1]Cunningham C,Hikima J,Jenny M J,et al.New resources for marine genomics:bacterial artificial chromosome libraries for the Eastern and Pacific oysters (Crassostrea virginicaandC.gigas)[J].Mar Biotechnol (NY),2006, 8(5):521-533.
[2]Hubert S,Hedgecock D.Linkage maps of microsatellite DNA markers for the Pacific oysterCrassostrea gigas[J].Genetics,2004, 168(1):351-362.
[3]Fleury E,Huvet A,Lelong C,et al.Generation and analysis of a 29,745 unique Expressed Sequence Tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database:the Gigas Database[J].Bmc Genomics,2009, 10:341.
[4]Hedgecock D,Lin J Z,DeCola S,et al.Transcriptomic analysis of growth heterosis in larval Pacific oysters(Crassostrea gigas)[J].Proc Natl Acad Sci U S A,2007,104(7):2313-2318.
[5]Wang Y,Guo X.Development and characterization of EST-SSR markers in the eastern oysterCrassostrea virginica[J].Mar Biotechnol (NY),2007, 9(4):500-511.
[6]Wang Y,Ren R,Yu Z.Bioinformatic mining of EST-SSR loci in the Pacific oyster,Crassostrea gigas[J].Anim Genet,2008, 39(3):287-289.
[7]Richard G F,Kerrest A,Dujon B.Comparative genomics and molecular dynamics of DNA repeats in eukaryotes[J].Microbiol Mol Biol Rev,2008, 72(4):686-727.
[8]Toth G,Gaspari Z,Jurka J.Microsatellites in different eukaryotic genomes:survey and analysis[J].Genome Res,2000, 10(7):967-981.
[9]Prasad M D,Muthulakshmi M,Madhu M, et al.Survey and analysis of microsatellites in the silkworm,Bombyx mori:frequency,distribution,mutations,marker potential and their conservation in heterologous species[J].Genetics,2005, 169(1):197-214.
[10]Subramanian S,Mishra R K, Singh L.Genome-wide analysis of microsatellite repeats in humans:their abundance and density in specific genomic regions[J].Genome Biol,2003, 4(2):R13.
[11]Huang X Q,Madan A.CAP3:A DNA sequence assembly program[J].Genome Research,1999, 9(9):868-877.
[12]Benson G.Tandem repeats finder:a program to analyze DNA sequences[J].Nucleic Acids Res,1999, 27(2):573-580.
[13]Canapa A,Barucca M,Cerioni P N,et al.A satellite DNA containing CENP-B box-like motifs is present in the antarctic scallopAdamussium colbecki[J].Gene,2000, 247(1-2):175-180.
[14]Lopez-Flores I,de la Herran R,Garrido-Ramos M A,et al.The molecular phylogeny of oysters based on a satellite DNA related to transposons[J].Gene,2004,339:181-188.
[15]Zhang L,Chen C,Cheng J,et al.Initial analysis of tandemly repetitive sequences in the genome of Zhikong scallop (Chlamys farreriJones et Preston)[J].DNA Seq,2008, 19(3):195-205.
[16]Li Y C,Korol A B,Fahima T,et al.Microsatellites:genomic distribution,putative functions and mutational mechanisms:a review[J].Mol Ecol,2002, 11(12):2453-2465.
[17]Zhang Z and Xue Q.Tri-nucleotide repeats and their association with genes in rice genome[J].Biosystems,2005, 82(3):248-256.
