关于药品再注册注射剂研究资料审评标准的探讨
2011-03-06段春改曹文利赵晨阳张伟琳
段春改 曹文利 赵晨阳 张伟琳
为加强药品注册管理,保障人体用药安全,按照国务院整顿和规范药品市场秩序专项行动的统一部署,自2007年起,国家局拟对核发的药品批准文号有效期满5年,需要继续生产的药品启动再注册工作。目前再注册的集中审评已经结束,成为日常性工作,但是在审评过程中遇到许多值得我们共同探讨的问题,尤其是安全性风险比较大的注射剂研究资料的审评工作,更值得我们共同商榷。根据安排,各省级药品监管部门将依照《药品管理法》等有关法律法规,结合药品批准文号清查、药品生产工艺和处方核查结果,开展药品再注册审查工作。河北省审评认证中心会同省局注册处、安监处、市场处、省药检所以及省内大型制药企业包括石药集团、华药集团和石家庄四药股份有限公司等先后召开专题研讨会或座谈会,并到企业中调研,最终综合考虑各方面要求,按照《化学药品注射剂基本技术要求(试行)》和《多组分生化药注射剂基本技术要求(试行)》(以下简称注射剂基本技术要求)进行注射剂研究资料的审评。
1 充分借鉴和依据前期工作
根据国家局要求,综合几次会议精神,以及调研情况,药品再注册审查工作充分结合药品批准文号清查、药品生产工艺和处方核查结果开展。注射剂的药品生产工艺和处方核查(以下简称工艺核查)是注射剂审查的一项必要依据,要求资料中必须附安监处的核查结果。核查结果为“继续生产”,予以再注册;核查结果为“继续生产,限期申报”,企业应提供申报补充申请的相关资料,予以再注册。未进行工艺核查,根据国家局在随后的【2010】168号发函,对未进行工艺核查或核查结果为“责令停产”的,待核查或复查符合要求后,予以再注册。在审评过程发现,企业容易缺失限期申报要求完成工作的资料,如原料供应商的变更等。工艺核查是注射剂审查的一项必要依据。
2 区分再注册与新药注册的不同
依据注射剂基本技术要求,充分考虑再注册不同于新药注册,作为老品种各方面技术已经比较成熟,所附研究资料与新药的要求有很大区别,一般要求做综述性陈述,或说明目前控制保障状态。审查的要点为:剂型、规格选择的必要性、合理性;注射剂原、辅料质量控制及来源;注射剂处方及制备工艺合理性、可行性研究,特别是灭菌工艺的验证研究等;注射剂质量研究;注射剂稳定性研究。
2.1 剂型、规格的合理性、必要性 此项不再要求提供完整的药学、药理以及临床研究资料,只要求综述性陈述本品种的剂型、规格的合理性和必要性。列出该品种剂型、规格,根据说明书中规定的用法用量,从方便临床用药、满足临床用药需要的角度加以阐述说明。但是一些企业却容易漏失,没有此项内容,直接从工艺验证资料开始。
2.2 原、辅料药质量控制及来源 本项需要按照相应项下要求说明目前的控制和保障状态,提供原、辅料药合法来源及质量控制的详细资料,如果有原料来源变更请附相应补充申请。(1)原料药质量控制:采用新研制的原料药与制剂同时申报的,应按照注射用原料药的要求提供规范完整的申报研究资料。有注射用原料药上市的,应使用有合法来源的注射用原料药。提供原料药合法来源及质量控制的详细资料,包括生产企业、批准文号、执行的质量标准等。若为进口原料药,还应提供进口注册证。若尚无注射用原料药上市,需对原料药进行精制并制定内控标准,使其达到注射用的质量要求。(2)辅料质量控制:使用已批准上市的注射用辅料,应提供辅料来源及质量控制的详细资料,包括生产企业、执行的质量标准等,有批准文号的还应提供批准文号,进口辅料还应提供进口注册证。使用尚未批准供注射途经使用的辅料,对于注射剂中有使用依据,但尚无符合注射用标准产品生产或进口的辅料,可对非注射途经辅料进行精制使其符合注射用要求,并制定内控标准。
需要特别说明的是,如果尚无注射用原料药上市,需对原料药进行精制并制定内控标准,使其达到注射用的质量要求。在注册申请时,除提供相关的证明性文件外,应提供精制的工艺验证资料和质量控制资料。如本省的注射用维生素C就属于此类情况。
2.3 注射剂处方及制备工艺合理性、可行性研究,特别是灭菌工艺验证等。
处方组成:列出处方中主药及辅料成份,并写明各种辅料在处方中的作用,并分析辅料用量及与主药的相互作用。
制备工艺验证项要求提供工艺流程图(标明洁净区级别)、工艺描述、工艺验证资料、灭菌工艺验证资料。提供完整的制备工艺及工艺验证的结果,对工艺过程本身是否稳定,是否易于控制进行总结和评价。需要注意需要混粉的品种,在处方组成、工艺描述以及工艺验证中都要体现混粉并进行相应验证。
灭菌工艺验证工作可结合生产线验证一并进行。不同剂型灭菌工艺验证资料也不同。
大容量注射剂需要准备终端灭菌工艺验证资料。终端灭菌产品,灭菌条件一般达到F0≥8,均应保证产品灭菌后的SAL不大于10-6。灭菌工艺验证主要包括以下试验:灭菌前微生物污染水平测定,包括灭菌前产品中的污染菌及其耐热性的测定;热穿透试验;微生物挑战试验:所用生物指示剂的耐热性及数量应对灭菌工艺构成必要的挑战,生物指示剂的耐热性应大于产品中常见污染菌的耐热性。采用过度杀灭法(F0≥12)灭菌工艺的,可不进行微生物挑战试验。
冻干粉针剂常规工艺验证包括培养基模拟灌装验证和除菌过滤系统适应性验证资料。培养基模拟灌装验证试验:至少在线灌装三批,每批的批量详见表1,判断该试验是否合格的标准见表1。应进行阳性对照试验及阴性对照试验。除菌过滤系统适应性验证试验:包括过滤系统相容性测试、过滤前后滤膜完整性测试,必要时尚需进行滤膜的微生物截留量测试。无菌分装粉针剂工艺验证工作主要为培养基灌装验证试验。灌装的批数、批量与合格标准见表1。
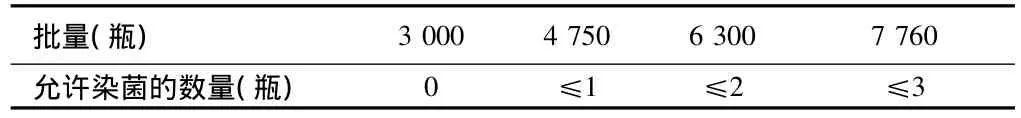
表1 培养基灌装试验的批量与判断合格的标准
小容量注射剂灭菌工艺验证资料因工艺的区别与大容量注射剂或冻干粉针的要求一致。需要注意的是,国家局在《关于做好药品再注册审查审批工作的补充通知》食药监注【2010】394号中,进一步加强了无菌保障水平F0值﹤8的品种无菌保障,要求应至少提交无菌工艺验证研究资料。因此这些品种如有充分的依据证明不能采用终端灭菌工艺,且为临床必需注射给药的品种,可考虑采用无菌生产工艺,相关技术要求同冻干粉针剂。对于过滤除菌工艺同时采用了流通蒸汽辅助灭菌的品种,建议修改为终端灭菌工艺,技术要求同大容量注射剂;对确实无法采用终端灭菌工艺的品种,应修改为无菌生产工艺,技术要求同冻干粉针剂。对于采用无菌生产工艺生产的小容量注射剂,生产线的验证应结合无菌生产工艺进行。
生化药参照以上相应剂型进行。注射剂基本技术要求特别提到,由于生化药的特殊性,需关注灭菌处理对产品质量/疗效以及产品安全性的影响。生化药的原材料来源于不同的动物组织或者体液,为保持特定/有效成分的生物活性,通常需要采用比较温和的生产工艺和提取制备条件,因而污染的潜在病毒可能未得到有效灭活/去除,给用药人群带来感染的风险性。因此,必须确立在生产工艺中包含能够有效灭活/去除病毒的特定工艺步骤,并验证其灭活/去除病毒的效能,提供病毒灭活/去除有效性验证资料。以保证制品的病毒安全性。
2.4 化学药品注射剂质量研究 请附现执行质量标准;检验方法验证至少应提供无菌检验方法的验证。
2.5 化学药品注射剂稳定性研究 注射剂稳定性研究的样品批次和规模、包装和放置条件和考察时间点参见《化学药物稳定性研究技术指导原则》,考察项目通常应包括性状、pH值/酸碱度、澄清度与颜色(或溶液的澄清度与颜色)、有关物质、干燥失重/水分(注射用粉末)、细菌内毒素/热原、无菌、不溶性微粒、可见异物、含量(效价)测定。注射剂稳定性研究内容包括影响因素试验、加速试验和长期试验,具体内容参见《化学药物稳定性研究技术指导原则》。报告中应包括稳定性研究结果评价。
3 专家审评把关注射剂审评
为了进一步做好注射剂的再注册工作,经过分析研究,化学药品和多组分化药注射剂需要通过组织专家技术审评通过后,方予以注册。审评专家从专家库中随机抽取决定。专家库由本省科研单位、药品检验单位、疾病控制中心以及省内大型生产单位的学术带头人组成。以此保证了注射剂审评质量。
总之,药品再注册是一项重要工作任务,当前我国药品标准水平还不能完全适应监管工作的需要,药监部门将把党中央提出的“用最严格的药品标准监督药品安全”的要求落实到具体工作中,围绕提高药品质量、保障用药安全的总体要求,进一步规范药品审评行为,严把药品审批关。
