植物来源的抗肿瘤药物研究进展
2011-02-02王超磊孙炳峰姚和权吴晓明徐进宜
王超磊,孙炳峰,姚和权,吴晓明,徐进宜*
(1.中国药科大学药学院,江苏南京 210009;2.中国科学院上海有机化学研究所,上海 200032)
植物来源的抗肿瘤药物研究进展
王超磊1,2,孙炳峰2,姚和权1,吴晓明1,徐进宜1*
(1.中国药科大学药学院,江苏南京 210009;2.中国科学院上海有机化学研究所,上海 200032)
综述喜树碱、紫杉醇、鬼臼毒素和combretastatin A-4等4类植物来源的抗肿瘤药物的作用机制、构效关系及衍生物的研究进展。植物来源的抗肿瘤药物已逐步在临床肿瘤治疗领域占据主导地位,而充分利用我国丰富的药用植物资源,研发高效低毒的天然抗肿瘤药物也已成为广大药学工作者的热点课题。
天然活性产物;结构修饰;衍生物;抗肿瘤活性;作用机制;构效关系
恶性肿瘤是危害人类生命和健康的严重疾病之一。从植物中寻找有效的抗肿瘤药物已成为国内外药学研究者的热点研究课题,从1981年到2008年的27年间已上市的抗肿瘤药物中,有62.9%来自天然产物或其衍生物[1-2],可见天然产物在抗肿瘤药物的研究与开发中的重要地位。目前,临床上已经筛选出20多种植物来源的抗肿瘤药物。
1 喜树碱类药物
1.1 抗肿瘤作用机制
DNA拓扑异构酶(Topo)是广泛存在于生物体内的一类必需酶,通过调节DNA超螺旋、连锁、去连锁以及核酸解链作用而影响DNA拓扑结构,主要分为TopoⅠ和TopoⅡ,其中TopoⅠ已成为设计新型抗肿瘤药物的重要靶酶。与TopoⅡ抑制剂相比,TopoⅠ抑制剂的抗肿瘤活性更高,抗瘤谱更广。研究表明:喜树碱(camptothecin,CPT,1)作为TopoⅠ抑制剂,并非通过抑制TopoⅠ的催化活性而发挥抗癌作用,而是通过与TopoⅠ-DNA可裂解复合物可逆性结合,形成CPT-TopoⅠ-DNA三元复合物,促进可裂解复合物的稳定,形成“路障”(road blocker),抑制复制叉(replication fork)的进程,从而导致细胞死亡[3]。
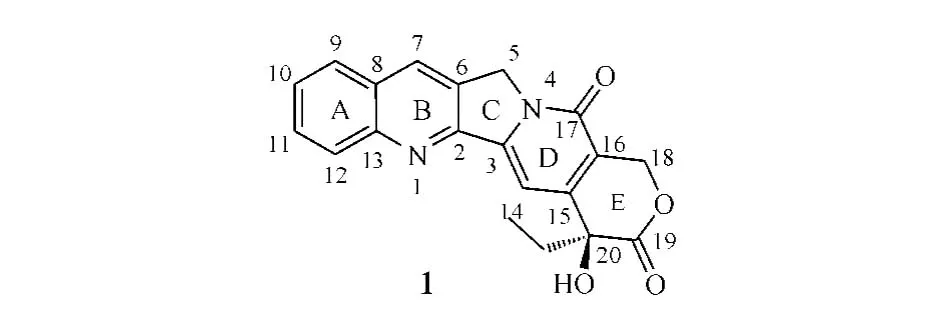
1.2 构效关系
CPT及其类似物是目前临床上广泛应用的一类特异性TopoⅠ抑制剂。喜树碱最早于1966年从珙桐科植物喜树(Camptothecaacuminate)根皮中分离出来,具有喹啉类生物碱内酯结构,A~D环的修饰对其活性会产生一定影响,而E环内酯环对其键合TopoⅠ更为必要,将E环水解开环或去除都会导致其抗癌活性完全消失。天然CPT水溶性较差,E环在生理条件下易开环而失去活性,因此其结构改造研究主要集中在增加其水溶性和E环稳定性方面。
研究发现,CPT及其衍生物结构中取代基的种类、数量和位置都会对其细胞毒活性产生影响。通常A环和B环(喹啉环)的修饰对化合物活性影响较小,7、9、11位引入合适的取代基可保持或提高化合物的活性并降低其毒性;C、D环的修饰或在12、14位引入取代基可使化合物活性降低或失去;在7位取代基上引入可形成氢键的基团能增强化合物与TopoⅠ的结合能力,从而提高其活性;E环只能耐受一些微小的修饰,例如,将六元内酯环扩环成七元内酯环,以提高内酯环稳定性及其化合物活性,而将20位羟基烷基化或酰化也可起到稳定内酯环及提高化合物活性的作用[4]。
1.3 结构修饰及相关衍生物
1.3.1 A环修饰喹啉环(A和B环)是CPT最常见的结构修饰位点,通过对其修饰改造,得到了许多水溶性较好、内酯稳定性增加、细胞毒活性较高的衍生物。目前已有3个此类CPT衍生物应用于临床,其中,在A环10位上引入亲水性基团以增加水溶性而得到的抗肿瘤前药伊立替康(irinotecan,2),先后于1994年和1996年在日本和美国获准上市,用于治疗结肠直肠肿瘤;在A环9和10位上分别引入亲水性基团叔胺基和羟基而得到的水溶性更高的拓扑替康(topotecan,3),于1996年被美国FDA批准上市,用作治疗卵巢肿瘤的二线药物,且其口服制剂于2007年在美国上市,用于治疗复发性小细胞肺癌。
在伊立替康开发成功不久,美国Supergen公司又在一系列A环取代的CPT衍生物中发现9-氨基CPT在体内外都表现出很高的细胞毒活性,但进一步的研究发现其半衰期较短,水溶性和脂溶性均较差,且对光、热、氧不稳定,给生产、储存带来困难,临床使用受到限制。因此,研发者将注意力转移到了合成9-氨基CPT时的中间体9-硝基CPT(rubitecan,4)上,其化学性质稳定,易获得,可在体内转化为活性形式——9-氨基CPT,该药早在20世纪90年代中期已完成治疗肺癌、乳腺癌、结肠直肠癌、卵巢癌、胃癌、前列腺癌、白血病和黑色素瘤等的Ⅰ、Ⅱ、Ⅲ期临床研究,目前该化合物用于胰腺癌治疗的研究仍处于Ⅲ期临床试验阶段[5]。

1.3.2 B环修饰在研究如何提高CPT类似物水溶性的同时,为了提高药物的血液稳定性及细胞摄取率,实现口服给药,人们又合成了一系列脂溶性CPT衍生物。其中,silatecans是B环7位连有硅烷基侧链的一类CPT衍生物,硅烷基侧链使得这类化合物具有很强的亲脂性,更易于分布到脂质双分子层中,避免被血浆中的酶快速降解,从而提高了生物利用度,改善了药动学性质。其最具代表性的化合物是karenitecin(BNP1350,5)和DB-67(6)[6],前者是美国BioNumerik公司开发的半合成CPT衍生物。用4种异种移植肿瘤的裸鼠模型进行的试验显示,karenitecin的生物利用度达67%,而拓扑替康为30% (Schellens等,BrJCancer,1996年)。另有试验表明:DB-67在SCID大鼠血浆中的半衰期为1.4 h,AUC为17 mg·h·L-1[7]。目前karenitecin治疗非小细胞肺癌(NSCLC)、卵巢癌、乳腺癌、结肠癌和黑色素瘤的研究已进入Ⅲ期临床试验阶段,用于治疗成人转移性实体肿瘤的研究也已进入Ⅰ期临床试验。
在B环7位上引入亲水基团而得到的水溶性CPT衍生物贝洛替康(belotecan,CKD-602,7)于2004年被韩国食品药品管理局批准上市,用于治疗卵巢癌和小细胞肺癌。

1.3.3 A和B环同时修饰鲁托替康(lurtotecan,8)即是由葛兰素史克公司开发的A和B环同时修饰的全合成水溶性CPT衍生物,对卵巢癌、乳腺癌和结肠癌等疗效显著。由于其7位引入了甲基哌嗪叔胺基团,10、11位引入了乙二氧基环,从而大大提高了水溶性和细胞毒活性。鲁托替康在pH为5时在水中最大溶解量达5.8 g·L-1,而CPT和拓扑替康分别为0.003和3.1 g·L-1。细胞毒性试验显示,鲁托替康的抗肿瘤活性为拓扑替康的3~5倍(Luzzio等,JMedChem,1995年);且临床前体内试验显示,一定剂量的鲁托替康可致裸鼠异种移植的HT29和SW48肿瘤缩小60%(Emerson等,CancerRes,1995年)。然而,Ⅰ期临床研究发现鲁托替康的疗效并不理想,进而开发了鲁托替康脂质体NX-211,目前NX-211治疗卵巢癌、头癌、颈癌和肺癌的研究已进入Ⅱ期临床试验阶段[8]。
依沙替康(exatecan,DX-895951f,9)是日本Daiichi公司开发的水溶性CPT类似物,它以一个带氨基的六元环相并合的结构将A和B环的7和9位连接,同时在A环10位引入甲基,11位引入氟原子,从而提高了E环内酯环的稳定性,水溶性也大大增加。临床前研究显示,依沙替康具有高抗癌活性,其抗乳腺癌、结肠癌、肾癌、胃癌、卵巢癌、肺癌和子宫癌的活性是拓扑替康的28倍(Mitsui等,JpnJ CancerRes,1995年)。目前,其治疗肉瘤、胆管癌和肺癌的Ⅰ、Ⅱ期临床试验正在进行中,且治疗局灶性和转移性胰腺癌的研究也已进入Ⅲ期临床试验[9]。
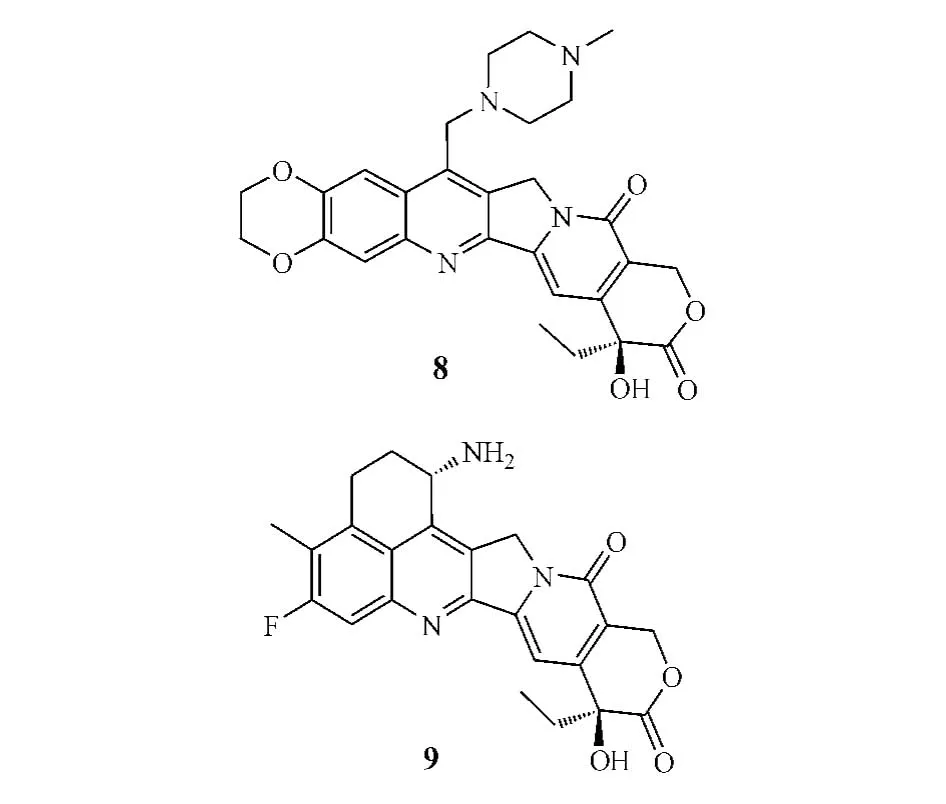
1.3.4 C和E环修饰DRF-1042(10)是由印度Dr.Reddy制药公司开发的、对C环修饰而得到的微水溶性半合成CPT衍生物,具良好的抗癌活性,临床前研究显示具较好的口服生物利用度和血浆稳定性[10]。Ⅰ期临床试验显示,该化合物半衰期为9.9 h,对难治性实体瘤疗效显著,并具有较好的口服药动学性质[11]。目前,该化合物正处于Ⅱ期临床研究阶段。
将六元内酯环扩大为3-羟基七元内酯环,即在羟基与羧基间引入亚甲基而获得的新型高喜树碱类化合物(homocamptothecins,hCPT,11),与CPT相比,其内酯环的稳定性大大增加,从而活性提高。在此类化合物中,具有代表性的化合物是Beaufour-Ipsen公司与Roche公司共同开发的diflomotecan (BN-80915,12),其在体外对HT29、A549和T24r肿瘤株的IC50分别达8.4、3.4和0.40 nmol·L-1,活性优于hCPT和CPT[12]。由于该化合物可与DNATopoⅠ形成三元不可裂解复合物,因此体内半衰期延长。目前,该化合物已进入Ⅱ期临床研究,有望成为第一个上市的hCPT类Topo抑制剂[13]。

2 紫杉醇类药物
2.1 抗肿瘤作用机制
正常情况下,微管和微管蛋白二聚体之间存在动态平衡,有丝分裂时微管在钙离子的作用下解聚,形成纺缍体和纺缍丝,牵引染色体向两极移动。紫杉醇(paclitaxel,Taxol®,13)主要与β微管蛋白N端第31位氨基酸和第217~231位氨基酸结合,促进微管蛋白二聚体的形成和微管装配,打破微管聚合与解聚的动态平衡,阻碍纺缍丝的形成,导致细胞周期停滞于G2/M期,使快速分裂的肿瘤细胞生长受抑并死亡。
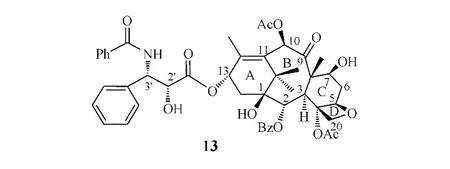
2.2 构效关系
紫杉醇是1971年由Wani等首先从短叶红豆杉中提取分离出来的具抗癌活性的二萜类化合物,1992年由美国FDA正式批准上市,用于治疗晚期卵巢癌。目前紫杉醇已作为一线抗肿瘤药物在40余国上市,而其半合成类似物多西他赛(docetaxel,Taxotere®,14)于1995年率先在墨西哥、南非上市,1998年在美国上市。虽然紫杉醇和多西他赛在临床上得到了广泛应用,但仍然有较多问题需要解决,例如水溶性较差以及易产生多药耐药性等。紫杉醇和多西他赛的多药耐药性的产生主要是由于它们易与P-糖蛋白结合而被代谢。因此,为了克服或部分克服这些缺点,研究者们对紫杉醇进行了一系列的结构改造,开发出多种新化合物实体。

紫杉醇13α位侧链的空间分布及构象对其活性至关重要:其中2'R/3'S的立体构型和3'位的酰胺取代都是其活性所必需,若3'位酰胺被氨基取代则活性消失;而3'位苯基或苯基类似基团亦为活性所必需;另外,2'位游离羟基也是活性必需基团,羟基酰化后,其体外活性降低,但体内活性未受影响。
紫杉醇骨架的[6/8/6]稠合方式对其活性十分重要,若发生改变或开环,其促进微管聚合的作用降低,细胞毒性完全丧失;4、5、20位的四元氧环也与其活性密切相关,若无此环或开环则活性完全丧失; 1位羟基缺失,其活性下降;2位苯甲酰氧基和4位乙酰氧基的存在对其活性至关重要,若失去则活性大大下降;7、9、10位含氧基团的存在对其活性影响不大,但若10位氧化成酮则活性消失[14]。
2.3 结构修饰及相关衍生物
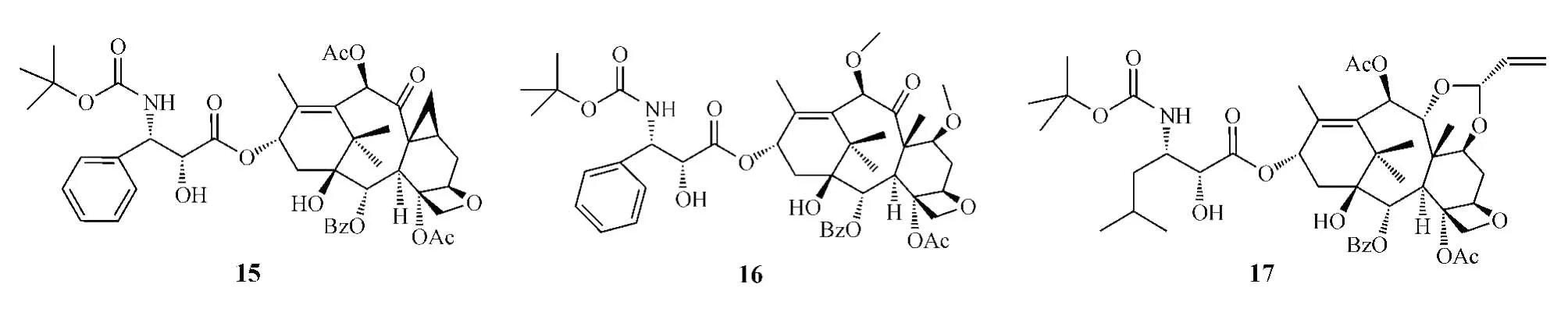
Larotaxel dehydrate(15)是由Sanofi-Aventis公司开发的紫杉醇衍生物,其8位甲基以环丙基替代,侧链中苯甲酰基用叔丁氧羰基取代。它对耐多西他赛的白血病细胞系p-388有细胞毒作用,并可穿过血脑屏障,这可能是其对P-糖蛋白的亲和力降低所致。Ⅱ期临床研究显示该化合物对耐紫杉醇的晚期乳腺癌有较高的治疗指数[15],目前其正处于Ⅲ期临床研究阶段。
Cabazitaxel(16)是由Sanofi-Aventis公司开发的多西他赛二甲氧基衍生物,即在多西他赛的7和10位羟基上引入两个甲基修饰而成,具有不被P-糖蛋白转运外排、可穿过血脑屏障的优点[16],其与泼尼松联用治疗激素抵抗性前列腺癌的研究正处于Ⅲ期临床试验阶段。
TPI-287(17)则是将紫杉醇侧链上3'位苯基替换为异丁基,并在母核上引入了一个丙烯基缩醛结构,导致其与紫杉醇及多西他赛在结构上发生较大变化,从而使其难与P-糖蛋白相结合,避免了P-糖蛋白介导的药物外排,克服了紫杉醇的多药耐药性。目前,该化合物处于Ⅰ/Ⅱ期临床研究阶段,用于治疗晚期胰腺癌和激素抵抗性前列腺癌[17]。
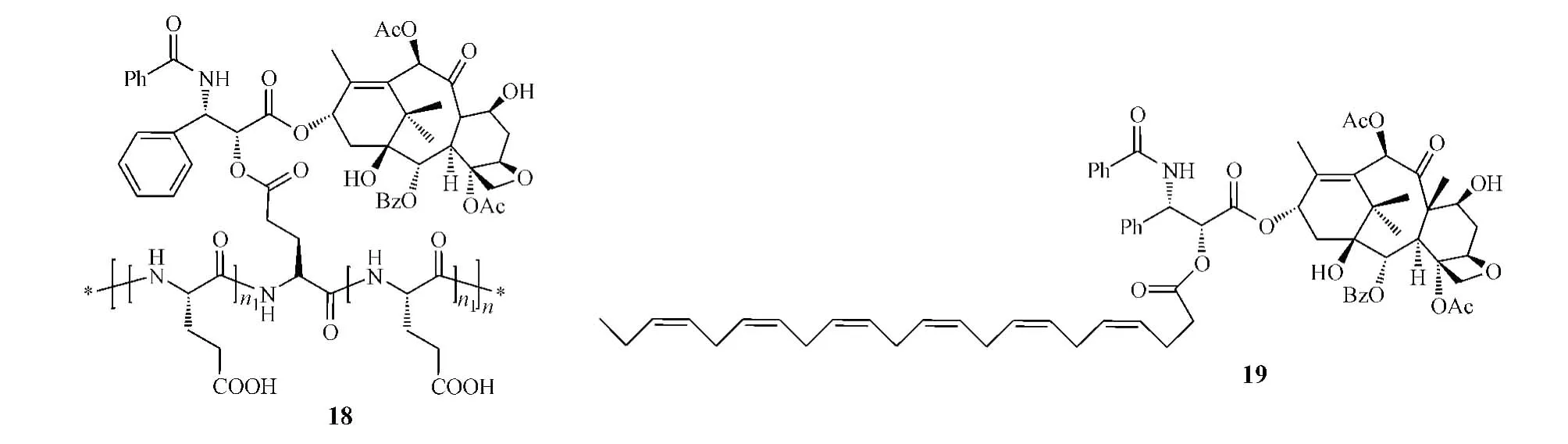
为提高紫杉醇的水溶性,改善其药动学性质,有研究者将紫杉醇和水溶性高分子聚合物聚谷氨酸通过酯键连接而形成聚谷氨酸-紫杉醇偶联物Xyotax (paclitaxel poliglumex,18)[18],该偶联物为一前药,可通过内吞作用进入细胞,在相关酶的作用下释放出游离的紫杉醇而发挥抗肿瘤作用,且由于其较大的结构,不能被P-糖蛋白识别而避免了P-糖蛋白介导的药物外排[19],同时由于其水溶性增加,避免了传统紫杉醇药物因为需使用表面活性剂而导致的一些副作用。目前,该前药处于Ⅲ期临床研究阶段。
研究表明:由于癌细胞代谢速率加快,其对脂肪酸摄取量增加,因此将脂肪酸与化疗药物共价连接,可提高药物对癌细胞的靶向性及治疗指数。Taxoprexin(DHA-paclitaxel,19)便是紫杉醇和二十二烷六烯酸(DHA)通过酯键连接而形成的DHA-紫杉醇偶联物[20]。临床前研究显示,该偶联物在体外完全没有活性,在血浆中转化为活性紫杉醇的速率很低,而在癌细胞中的转化率是血浆中的21倍,提高了药物靶向性。在移植M109肿瘤小鼠中进行的实验显示,分别给予等摩尔剂量或等毒性剂量的该偶联物和紫杉醇后,该偶联物的AUC分别是紫杉醇的8倍或57倍[21]。目前,该偶联物处于Ⅲ期临床研究阶段。
3 鬼臼毒素类药物
3.1 抗肿瘤作用机制
研究表明:鬼臼毒素(podophyllotoxin,20)及其衍生物由于结构的不同主要有两种抗肿瘤作用机制:鬼臼毒素本身可通过破坏有丝分裂的细胞中微管蛋白集结以及微管的形成,使细胞有丝分裂停滞在M期,干扰肿瘤细胞分裂,从而抑制肿瘤生长;而依托泊苷和替尼泊苷等鬼臼毒素的4'位去甲基衍生物则是以共价键形成稳定的药物-DNA-TopoⅡ三元复合物,最终导致DNA单链及双链的断裂,使肿瘤细胞周期终止于G期(DNA合成前期)[22]。尽管鬼臼毒素及其衍生物与微管蛋白或TopoⅡ相互作用的方式尚不明确,但作为TopoⅡ抑制剂的鬼臼毒素衍生物与抑制微管蛋白聚合的衍生物在结构上的差异已比较明确,主要表现在TopoⅡ抑制剂衍生物的4'位甲氧基脱甲基成羟基、4位取代基为β构型以及4位上有大体积的取代基团。

3.2 构效关系
鬼臼毒素是从小蘖科鬼臼属植物中提取到的木质素类抗肿瘤活性成分,具有显著的抗肿瘤活性,但毒副作用也较大,尤其是对胃肠道的毒性较大,不适于临床使用。为了提高其细胞毒活性和TopoⅡ抑制活性,降低毒副作用,人们尝试对其A、C、D、E环进行广泛修饰,合成了大量衍生物。现已发现对A环的修饰往往会造成化合物活性降低,因此目前进入临床应用及临床研究的鬼臼毒素衍生物均为C、E环修饰产物,且大多为TopoⅡ抑制剂。
研究表明:作为TopoⅡ抑制剂的鬼臼毒素衍生物,其C环4位取代基必须为β构型,若为α构型则活性显著降低;4'位羟基为活性所必需;D环反式γ-内酯环是其具有强效抗肿瘤活性的一个重要结构[23],但近年来,也有诸多文献报道了具抗肿瘤活性的D环开环的鬼臼毒素衍生物[24-26];此外,A环间二氧戊环以及E环也是活性必需结构。
3.3 结构修饰及相关衍生物
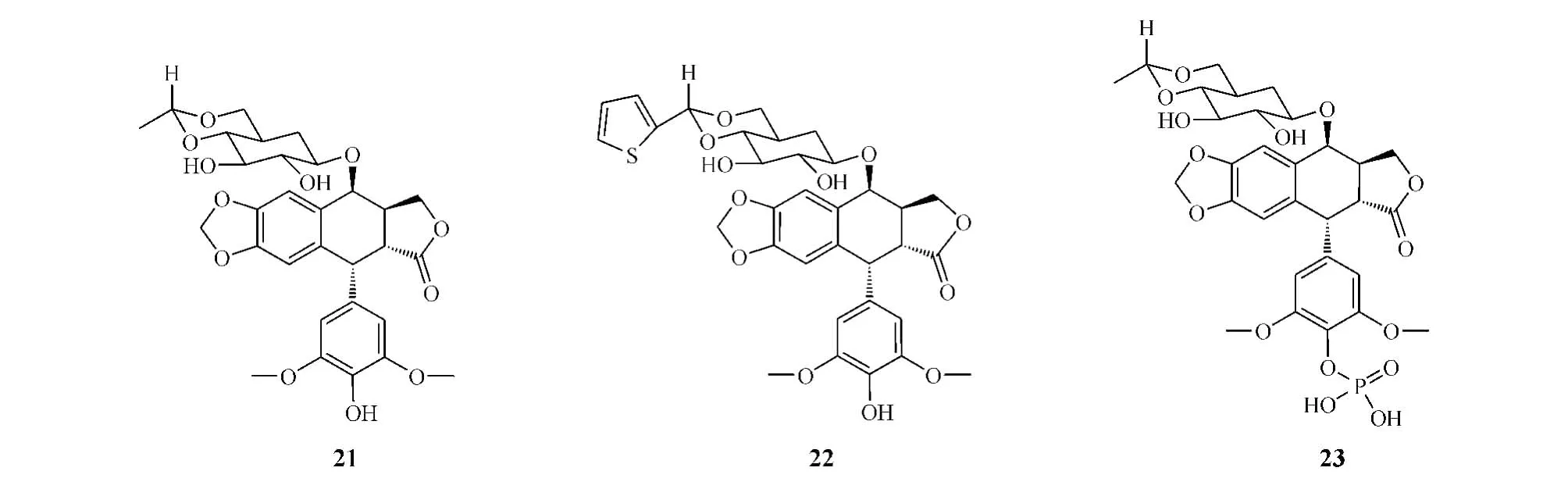
早在20世纪60年代,瑞士Sandoz公司先后半合成了两个鬼臼毒素的糖苷化合物,即依托泊苷(etoposide,21)和替尼泊苷(teniposide,22),它们均为C、E环修饰的衍生物,具有高抗肿瘤活性,均被美国FDA批准上市,广泛用于睾丸癌、淋巴癌、白血病、NSCLC等的临床治疗,且依托泊苷的水溶性前药、注射用磷酸依托泊苷(etopopohos,23)也获准上市。然而,这类化合物存在水溶性差及易产生获得性耐药、骨髓抑制和严重的胃肠道反应等缺点[27]。因此,寻找高效低毒、对多药耐药肿瘤细胞有效的鬼臼毒素类抗肿瘤药物已成为当前研究热点。
由日本Nippon Kayaku公司研发的NK-611 (24)是用一个二甲胺基取代依托泊苷C环上乙叉糖苷部分的2″位羟基而获得的鬼臼毒素衍生物,二甲氨基的引入使得其水溶性显著提高,达依托泊苷的120倍。研究人员发现,NK-611对TopoⅡ有很强的抑制作用,对多种人类癌细胞系,包括肺癌、胃肠道癌、卵巢癌、辜丸癌、乳腺癌、头颈癌和白血病等都有很好的细胞毒活性,且对人类肿瘤异种移植模型也有较高抗肿瘤活性。其药效与依托泊苷相当,但依托泊苷和NK-611会产生交叉耐药性。目前,该化合物已进入Ⅱ期临床研究阶段(Rassmann等,InvestNewDrugs,1999年)。
GL-331(25)是在C环上引入4-硝基苯胺基、E环脱去一个甲基而得到的鬼臼毒素衍生物,与依托泊苷相比,其在体内外实验中都表现出较强的抗肿瘤活性。对KB细胞系的细胞毒活性测试显示,依托泊苷和GL-331对肿瘤细胞的ID50分别为0.49和0.2μmol·L-1,而对TopoⅡ的ID50分别为50和10μmol·L-1(Wang等,JMedChem,1990年)。用鼻咽癌、结肠癌、肝癌、胃癌、宫颈癌和结肠癌细胞进行的试验显示,GL-331对这些癌细胞的ID50在0.5~2μmol·L-1之间,是依托泊苷的3~10倍(Huang等,CancerLett,1996年)。该化合物目前已进入Ⅱ期临床研究阶段,主要用于耐依托泊苷的恶性肿瘤的治疗[28]。
TOP-53(26)是在C环上引入了一个乙基二叔胺基团而获得的鬼臼毒素衍生物,与依托泊苷相比,是一个更有效的TopoⅡ抑制剂,对TopoⅡ的抑制活性是依托泊苷的2倍(IC50分别为59和106μmol·L-1);且在细胞模型和动物模型中,对多种不同肿瘤细胞系都有较好的抑制活性,尤其对NSCLC的疗效更好(Utsugi等,CancerRes,1996年)。研究发现,TOP-53能特异性地分布到肺部,因而更适用于肺癌的治疗[29]。目前,该化合物正处于Ⅱ期临床研究阶段。
4 Combretastatin A-4类药物
4.1 抗肿瘤作用机制
Combretastatins是Pettit小组从南非植物Combretumcaffrum中提取分离的具有抗肿瘤活性的系列天然二苯乙烯类化合物,体外试验表明:其具有较高的抑制肿瘤细胞增殖活性,其中combretastatin A-4(CA-4,27)的活性最强,它不仅对肿瘤细胞(包括多药耐药肿瘤细胞)的增殖具有抑制活性,而且还可有效抑制微管蛋白聚合以及选择性抑制肿瘤血管增生。
研究显示,CA-4能够竞争性抑制微管与秋水仙碱的结合,表明CA-4具有与秋水仙碱相似的微管结合位点,可致肿瘤细胞和内皮细胞周期阻滞于G2/M期。CA-4的抗肿瘤血管作用机制与血管生成抑制剂不同,后者仅阻止新血管的生成,而CA-4则可特异性靶向破坏已生成的肿瘤血管,使肿瘤缺失氧气和营养供给,从而导致肿瘤“饿死”[30]。

4.2 构效关系
CA-4虽然在体外实验中表现出良好的抗癌活性,但因水溶性差,且顺式二苯乙烯结构不稳定,易异构化成反式构型,其体内活性并不能令人满意。因此,药物化学家们对CA-4进行结构修饰,合成了数百个CA-4衍生物。由于CA-4结构简单,且A环的修饰往往导致化合物活性降低,故对其修饰主要围绕B环进行,目前进入临床研究的CA-4衍生物大多是B环修饰产物。
研究表明:三甲氧基苯环(A环)是CA-4类化合物具有细胞毒性和抑制微管蛋白活性所必需的,这可能是由于3个甲氧基在空间结构上更有利于化合物与作用位点结合;保持CA-4衍生物的顺式结构和A、B两环间存在刚性基团,是化合物获得较高的细胞毒性和微管蛋白抑制活性的必要保证,因为这样可使A、B两环分别与微管蛋白的α和β链上两处结合位点的作用更加紧密;B环的4位甲氧基也可用一些小分子的取代基,如甲基、卤素等取代,而对B环羟基的修饰可形成CA-4前药,增强疗效[31]。
4.3 结构修饰及相关衍生物
将CA-4的B环上3位羟基磷酸化而得到的高水溶性前药CA-4磷酸二钠盐(CA-4P,28)能有效抑制微管蛋白的聚合,IC50为(214±1.4)μmol·L-1(Lin等,MolPharmacol,1988年)。体内研究显示,给原位和皮下移植实体瘤的小鼠腹腔注射CA-4P 100 mg·kg-1(300 mg·m-2)6 h后,小鼠肿瘤血管容积减少90%,肿瘤血流减少50%~60%(Chaplin等,AnticancerRes,1999年)。由于在临床前研究中显示出较明显的体内外抗肿瘤作用,CA-4P已于2002年进入临床研究,并于2006年5月被美国FDA批准为罕见病药物,用于治疗卵巢癌患者,尤其是与卡铂和紫杉醇联用,治疗对铂类药物产生耐药性的患者及未分化、髓样、Ⅳ期乳头状和Ⅳ期滤泡型甲状腺癌患者。该药也在欧洲获得了罕见病药物资格,用于治疗未分化甲状腺癌患者。其用于治疗不同肿瘤的研究目前在欧美正处Ⅰ/Ⅱ/Ⅲ期临床试验阶段[32-33]。
对天然产物combretastatin A-1(CA-1,29)进行结构修饰而获得的二磷酸衍生物OXi4503(30)由美国OXiGENE公司研制。临床前研究显示,该药对CaNT乳腺癌鼠模型的疗效显著,其腹腔注射1 mg· kg-1后,肿瘤血管容积减少50%;当剂量加大到10、25、50 mg·kg-1时,肿瘤血管容积减少80%以上;当给药剂量达100、200和400 mg·kg-1时,肿瘤细胞生长明显受抑[34]。OXi4503目前正处Ⅰ期临床研究阶段。
Ombrabulin(AVE8062,31)是Sanofi-Aventis公司研发的水溶性CA-4前药,即CA-4的B环上3位羟基被生物电子等排体胺基取代而获得的衍生物,其在水中的最大溶解量是14.7 g·L-1,高于CA-4的0.11 g·L-1(Ohsumi等,AnticancerDrugDes,1999年)。动物实验显示,该药能抑制肿瘤增殖,在近40种肿瘤动物模型中,其静脉注射给药后24 h内,均观察到肿瘤发生缺血性坏死[35];且在给药30 min后,肿瘤血流完全停滞,而正常组织血流仅少量减少,并在24 h内即恢复至给药前水平[36]。目前,该药已进入Ⅲ期临床研究,用于治疗NSCLC[37]。

5 结语
从植物中寻找开发有效抗肿瘤新药的前景是广阔的。目前,众多不同结构和作用机制的天然活性产物已成为国内外抗肿瘤新药研究的热点,如冬凌草甲素(oridonin)、白桦酸(betulinic acid)、23-羟基白桦酸(23-hydroxy betulinic acid)等萜类化合物,由于其独特的结构和良好的抗肿瘤活性,正引起越来越多的药学工作者的重视,其中白桦酸在国内作为抗黑色素瘤药物正处于Ⅰ期临床研究阶段,冬凌草甲素正处在申报新药阶段。笔者所在课题组对23-羟基白桦酸以及冬凌草甲素的结构修饰研究也取得一定进展,已初步获得这些化合物的构效关系,并看好它们的进一步研究价值和应用前景[38-42]。
在我国连续启动的两批重大新药创制项目中,植物来源药物特别是利用我国植物资源的天然药物的研发都被列为重点支持对象。由于植物来源的抗肿瘤药物的作用机制独特,抗癌疗效显著,已在临床应用上逐步占据主导地位。但是这些药物也存在选择性差,易产生毒副作用及耐药性,甚至杀死正常细胞等问题。因此,在从天然产物中寻找安全、经济和高选择性的抗癌活性物的基础上,对这些植物来源的化合物进行结构修饰,以获得低毒性、高活性的抗肿瘤候选新药,仍然是今后药学研究者的努力方向。有理由相信,随着人们对天然产物研究的深入,从天然产物中提取有效成分,并通过对其进行结构简化和修饰,必将会有更多的抗肿瘤活性成分被研发出来,并应用于临床。
[1]Cragg G M,Newman D J.Impact of natural products on developing new anti-cancer agents[J].Chem Rev,2009,109(7):3012-3043.
[2]Mohammad S.Anticancer agents from medicinal plants[J].Bangladesh J Pharmacol,2006,1(2):35-41.
[3]Liu L F,Desai SD,Li T K,et al.Mechanism of action of camptothecin[J].Ann NY Acad Sci,2000,922(1):1-10.
[4]Li Q Y,Zu Y G,Shi R Z,et al.Review camptothecin:current perspectives[J].Curr Med Chem,2006,13(17): 2021-2039.
[5]Liew ST,Yang L X.Design,synthesis and developmentof novel camptothecin drugs[J].Curr Pharm Des,2008,14 (11):1078-1097.
[6]Basili S,Moro S.Novel camptothecin derivatives as topoisomerase I inhibitors[J].Expert Opin Ther Patents,2009,19(5):555-574.
[7]ZamboniW C,Jung L L,Strychor S,et al.Plasma and tissue disposition of nonliposomal DB-67 and liposomal DB-67 in C.B-17 SCID mice[J].Invest New Drugs,2008,26 (5):399-406.
[8]Flaherty K T,Stevenson JP,Twelves C J,et al.The clinical development of lurtotecan:experience with water-soluble and liposomal forms[M]∥Adams Val R,Burke T G.Cancer Drug Discovery and Development:Camptothecins in Cancer Therapy(PartⅡ).Totowa:Humana press,2005:301-316.
[9]Abou-Alfa G K,Letourneau R,Harker G,etal.Randomized PhaseⅢstudy of exatecan and gemcitabine compared with gemcitabine alone in untreated dvanced pancreatic cancer[J].J Clin Oncol,2006,24(27): 4441-4447.
[10]Upreti V V,Mamidi R N,Katneni K,et al.Quantitative determination of DRF-1042 in human plasma by HPLC: validation and application in clinical pharmacokinetics[J].Biomed Chromatogr,2003,17(6):385-390.
[11]Chatterjee A,Digumarti R,Katneni K,et al.Safety,tolerability,and pharmacokinetics of a capsule formulation of DRF-1042,a novel camptothecin analog,in refractory cancer patients in a bridging phase I study[J].J Clin Pharmacol,2005,45(4):453-460.
[12]Lavergne O,Demarquay D,Bailly C,et al.Topoisomerase I-mediated antiproliferative activity of enantiomerically pure fluorinated homocamptothecins[J].J Med Chem,2000,43(11):2285-2289.
[13]Kroep JR,Gelderblom H.Diflomotecan,a promising homocamptothecin for cancer therapy[J].Expert Opin Investig Drugs,2009,18(1):69-75.
[14]FangW S,Liang XT.Recentprogress in structure activity relationship and mechanistic studies of taxol analogues[J].Mini Rev Med Chem,2005,5(1):1-12.
[15]Metzger-Filho O,Moulin C,Evandro A,et al.Larotaxel: broadening the road with new taxanes[J].Expert Opin Investig Drugs,2009,18(8):1183-1189.
[16]Tuma R S.Cabazitaxelmay be new second-line standard for patients with castrate-resistant prostate cancer[J].Oncol Times,2010,32(8):44-46.
[17]Chen SM,Meng L H,Ding J.New microtubule-inhibiting anticancer agents[J].Expert Opin Investig Drugs,2010,19(3):329-343.
[18]Bonomi P.Paclitaxel poliglumex(PPX,CT-2103):macromolecular medicine for advanced non-small-cell lung cancer[J].Expert Rev Anticancer Ther,2007,7(4): 415-422.
[19]Singer JW,Baker B,De Vries P,et al.Poly-(L)-glutamic acid-paclitaxel(CT-2103)[XYOTAX],a biodegradable polymeric drug conjugate:characterization,preclinical pharmacology,and preliminary clinical data[M]∥Maeda H,Kabanov A,Kataoka K,et al.Polymer Drugs in the Clinical Stage:Advantage and Prospects.New York:Kluwer Academic/Plenum Publishers,2003:81-99.
[20]Jones R J,Hawkins R E,Eatock M M,et al.A phase II open-label study of DHA-paclitaxel(Taxoprexin)by 2-h intravenous infusion in previously untreated patients with locally advanced ormetastatic gastric or oesophageal adenocarcinoma[J].Cancer Chemother Pharmacol,2008,61 (3):435-441.
[21]Bradley M O,Webb N L,Anthony FH,et al.Tumor targeting by covalent conjugation of a natural fatty acid to paclitaxel[J].Clin Cancer Res,2001,7(10):3229-3238.
[22]Srivastava V,Negi A S,Kumaar JK,etal.Plant-based anticancer molecules:a chemical and biological profile of some important leads[J].Bioorg Med Chem,2005,13 (21):5892-5908.
[23]Liu Y Q,Yang L,Tian X.Podophyllotoxin:current perspectives[J]Curr Bioact Compd,2007,3(1):37-66.
[24]Castro M A,Jose M,CorralM,etal.Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities[J].JMed Chem,2010,53(3):983-993.
[25]Gordaliza M,Castro M A,Miguel JM,et al.Synthesis and cytotoxic evaluation of C-9 oxidized podophyl-lotoxin derivatives[J].Bioorg Med Chem,2007,15(4): 1670-1678.
[26]Larsson O,Axelson M.Use of cyclolignans and new cyclolignans:US,7709526B2[P].2010-05-04.
[27]Mukherjee A K,Basu S,Sarkar N,et al.Advances in cancer therapy with plant based natural products[J].Curr Med Chem,2001,8(12):1467-1486.
[28]Liu JM,Chen L T,Chao Y,etal.Phase IIand pharmacokinetic study of GL331 in previously treated Chinese gastric cancer patients[J].Cancer Chemother Pharmacol,2002,49(5):425-428.
[29]Masahiko Y,Takashi K,Kumio A,etal.Specific distribution of TOP-53 to the lung and lung-localized tumor is determined by its interaction with phospholipids[J].Clin Cancer Res,2000,6(11):4396-4401.
[30]任萱,林莉萍,丁健.Combretastatin A4的抗肿瘤作用研究进展[J].中国新药杂志,2007,16(17): 1336-1340.
[31]Chaudhary A,Pandeya SN,Kumar P,et al.Combretastatin A-4 analogs as anticancer agents[J].Mini Rev Med Chem,2007,7(12):1186-1205.
[32]Rustin G J,Shreeves G,Nathan P D,et al.A Phase Ib trial of CA4P(combretastatin A-4 phosphate),carboplatin,and paclitaxel in patients with advanced cancer[J].Br JCancer,2010,102(9):1355-1360.
[33]Samarin J,Rehm M,Krueger B,et al.Up-regulation of connective tissue growth factor in endothelial cells by the microtubule-destabilizing agent combretastatin A-4[J].Mol Cancer Ther,2009,7(2):180-188.
[34]Hill S A,Tozer G M,Pettit G R,et al.Preclinical evaluation of the antitumour activity of the novel vascular targeting agent Oxi 4503[J].Anticancer Res,2002,22 (3):1453-1458.
[35]Hori K,Saito S.Microvascular mechanisms by which the combretastatin A-4 derivative AC7700(AVE8062) induces tumour blood flow stasis[J].Br J Cancer,2003,89(7):1334-1344.
[36]Hori K.Antineoplastic strategy:Irreversible tumor blood flow stasis induced by the combretastatin A-4 derivative AVE8062(AC7700)[J].Chemotherapy,2005,51(6): 357-360.
[37]Delmonte A,Sessa C.AVE8062:a new combretastatin derivative vascular disrupting agent[J].Expert Opin Investig Drugs,2009,18(10):1541-1548.
[38]Bi Y,Xu JY,Wu XM,et al.Synthesis and cytotoxic activity of 17-carboxylic acid modified 23-hydroxy betulinic acid ester derivatives[J].Bioorg Med Chem Lett,2007,17 (5):1475-1478.
[39]Xu JY,Yang JY,Ran Q,et al.Synthesis and biological evaluation of novel 1-O-and 14-O-derivatives of oridonin as potential anticancer drug candidates[J].Bioorg Med Chem Lett,2008,18(16):4741-4744.
[40]刘宏民,阎学斌,刘振中.冬凌草甲素衍生物及其制备方法:中国,99101179.1[P].2000-06-07.
[41]徐进宜,吴晓明,杨静怡,等.冬凌草甲素类衍生物、其制备方法及用途:中国,200710133915.X.[P].2010-06-02.
[42]Willmann M,Wacheck V,Buckley J,et al.Characterization of NVX-207,a novel betulinic acid-derived anticancer compound[J].Eur J Clin Invest,2009,39(5): 384-394.
Progresses in Study on the Plant-originated Anti-tumor Drugs
WANG Chao-lei1,2,SUN Bing-feng2,YAO He-quan1,WU Xiao-ming1,XU Jin-yi1
(1.SchoolofPharmacy,ChinaPharmaceuticalUniversity,Nanjing210009,China;2.ShanghaiInstituteof OrganicChemistry,ChineseAcademyofSciences,Shanghai200032,China)
The recent progresses in the mechanism of action and structure-activity relationship of four classes of the plant-originated anti-tumor drugs,camp to therins,paclitaxels,podophyllotoxins and combretastatin A-4,as well as their derivatives have been summerized in this paper.The plant-originated antitumor drugs have been gradually dominated in the clinical cancer treatment.Taking full advantage of rich resources of medicinal plants in China and developing new natural anti-tumor drugswith high activity and low toxicity have now become a hot topic studied by pharmaceutical researchers.
active natural product;structure modification;derivatives;anti-tumor activity;mechanism of action;structure-activity relationship
R979.1
A
1001-5094(2011)05-0193-10
10.3969/j.issn.1001-5094.2011.05.001
[接受日期]2011-03-03
[项目资助]国家自然科学基金项目(No.30973610);教育部科学技术研究重点项目(No.108069)
*通讯作者:徐进宜,教授;
研究方向:天然活性产物的结构修饰、半合成及生物活性研究;
Tel:025-83271445;E-mail:jinyixu@china.com
(责任编辑:范鸣)
