抗肿瘤药Palifosfamide tromethamine
2011-02-02杨臻峥
抗肿瘤药Palifosfamide tromethamine
palifosfamide;抗肿瘤药;软组织肉瘤
ZIOPHARM Oncology公司开发的新型抗肿瘤药palifosfamide(异磷酰胺氮芥)为经典抗肿瘤药异环磷酰胺的活性代谢物,是一种具有双重功能的烷化剂,可使DNA链间发生不可逆的交联,从而导致肿瘤细胞死亡。临床前研究显示,palifosfamide对荷有人类肉瘤或耐异环磷酰胺肿瘤的模型小鼠均有显著治疗作用,且安全性高于异环磷酰胺,因为palifosfamide与异环磷酰胺不同,不会代谢为具有膀胱毒性或中枢神经系统毒性的丙烯醛或氯乙醛;另外,palifosfamide可直接作用于肿瘤细胞,不需醛脱氢酶的激活,故有可能克服异环磷酰胺所致的耐药问题。然而,palifosfamide化学稳定性较差,不适合直接作为药物使用,故ZIOPHARM Oncology公司推出了palifosfamide的赖氨酸盐及氨丁三醇盐。
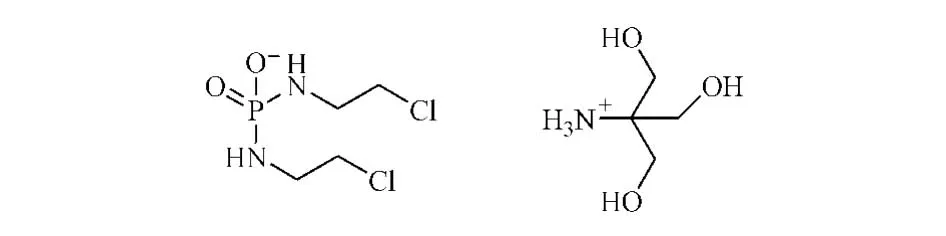
本品化学结构式:
CAS:31645-39-3
药理学研究体外研究显示,palifosfamide赖氨酸盐对人横纹肌肉瘤RD和RH30细胞株,尤文肉瘤(Ewing's sarcoma)SK-PN-DW 和 SK-ES-1 细胞株,骨肉瘤 Saos-2、OS222、OS29 和 OS230 细胞株及滑膜肉瘤HSSYII和SYOI细胞株均有显著细胞毒作用,除对 OS222细胞株的 IC50为 7 mg·L-1外,对其他所有受试细胞株的 IC50在0.5~1.5 mg·L-1之间。
多项临床前药理研究结果显示,palifosfamide对白血病或淋巴瘤细胞的体外活性及荷瘤小鼠体内肿瘤的抑制作用均为异环磷酰胺的10~30倍。此外,本品对荷有耐异环磷酰胺淋巴瘤小鼠的体内肿瘤也有显著的抑制活性。
研究人员采用荷有RH30和骨肉瘤细胞株(包括耐环磷酰胺的OS31株和对环磷酰胺敏感的OS33株)的SCID小鼠,对其在palifosfamide最大耐受剂量(MTD,100 mg·kg-1·d-1,iv,连续给药 3 天)下的无病生存期(EFS)进行了评价。结果显示,接受palifosfamide的荷瘤小鼠的EFS均比未用药组明显延长(P值分别为0.03、0.000 9和0.01)。
一项在荷有人乳腺癌MX-1细胞株的小鼠中进行的研究显示,静脉注射palifosfamide赖氨酸盐及palifosfamide的 MTD 分别为93 和40 mg·kg-1,相比于未给药组,给予上述两药的小鼠生存期分别延长了10.2和2.1天。此外,未见本品对肾脏、膀胱及中枢神经有毒副作用。
两项在荷有MX-1细胞株的小鼠中进行的研究结果显示,腹腔注射palifosfamide氨丁三醇盐的中毒剂量为 81 mg·kg-1·d-1,而 palifosfamide 赖氨酸盐在腹腔给药剂量为36mg·kg-1·d-1时即可观察到毒性。
经口使用palifosfamide氨丁三醇盐对荷有白血病P388细胞株和MX-1细胞株的模型小鼠均显示抗癌作用,且经口给药和胃肠外给药这2种给药方式在其各自的MTD下抗癌活性相近,提示本品具有开发为口服制剂的可行性。
此外,研究人员考察了palifosfamide氨丁三醇盐(54、24 或12 mg·kg-1·d-1,ip,连续给药 5 天)与多柔比星(8、5.3 或 3.5 mg·kg-1·d-1,iv,第 1、4 和8天给药)联用对荷有MX-1细胞株的小鼠的肿瘤抑制作用。结果显示,palifosfamide氨丁三醇盐给药剂量为 12 或 24 mg·kg-1·d-1、多柔比星剂量为8 mg·kg-1·d-1时,两药联用的肿瘤抑制活性明显强于单用其中任何一种(P<0.001);两药联用可被良好地耐受,未见小鼠体重减轻现象。另有一项研究结果显示,palifosfamide氨丁三醇盐与多西紫杉醇也有协同增效作用。
药动学与代谢药动学研究结果显示,给SD大鼠静脉注射 palifosfamide(40 mg·kg-1)后其以线性方式消除,平均半衰期(t1/2)为12.7 min,且符合单室动力学模型。本品总清除率平均值为(11.0± 4.4)mL·min-1[范围为 6.0 ~ 18.3 mL·min-1],分布容积平均值为(220±156)mL,血浆蛋白结合率为(55.1±0.1)%。本品在血浆和红细胞中的分配比为 4.9∶1。
研究人员还对palifosfamide氨丁三醇盐(20、30和40 mg·kg-1,po或iv)在雌性SD 大鼠中的药动学特性进行了考察。结果显示,本品在给药后0.5小时血药浓度达到峰值,t1/2范围为0.25~0.64 h。经口使用上述3个剂量下的生物利用度分别为48%、65%和73%,总体平均生物利用度为62%。在雄性大鼠中进行的药动学研究结果也与之基本相近,但平均生物利用度为41%。
安全性一项在小鼠中进行的安全性研究显示,palifosfamide氨丁三醇盐(iv,连续给药3天)的LD10和 LD50分别为 133 和 220 mg·kg-1,palifosfamide则分别为119和149 mg·kg-1。本品的剂量限制性毒性(DLT)为骨髓衰竭。
一项名为IPM1001的非随机、开标记的Ⅰ期临床研究对 palifosfamide 赖氨酸盐(30、42、59、83、116、162、227 和 318 mg·m-2,iv)在曾接受标准化疗后出现肿瘤转移和(或)无法手术切除的癌症患者中的安全性进行了评价。每3周为1个疗程,每个疗程给药3次或1次,共6个疗程,若出现DLT或疾病进展则停止治疗。结果显示,最常见的不良反应包括恶心、疲劳和厌食;未见骨髓毒性及明显的心脏、肝脏毒性。
一项名为IPM2001的单组、非随机、开标记的Ⅰ/Ⅱ期临床研究对palifosfamide赖氨酸盐(iv)在晚期肉瘤患者中的安全性进行了评价。每3周为1个疗程,每个疗程连续给药3天,每天1次,共6个疗程,若出现DLT或疾病进展则停止治疗。Ⅰ期研究中起初设定的剂量为590 mg·m-2,但研究人员发现该剂量可导致肾毒性,因而被降至413 mg·m-2,经证实,该剂量为MTD,亦被定为Ⅱ期研究中的剂量。此外,Ⅱ期研究中还包括一项对palifosfamide氨丁三醇盐(iv)的生物等效性评价。两项研究结果显示,最常见不良反应为恶心(44%)、低钾血(44%)、疲劳(32%)、贫血(34%)、低磷酸盐血(32%)、血浆碱性磷酸酶水平升高(32%)、血浆肌酐水平升高(24%)、显微镜血尿(24%)、呕吐(22%)以及低血钙或血浆乳酸脱氢酶水平升高(20%)。研究过程中未见明显心脏或肝脏毒性。
临床研究在一项由13名患有难治性晚期肿瘤的受试者(软组织肉瘤患者8名,小细胞肺癌、神经内分泌癌和骨肉瘤患者共5名)参加的Ⅰ期临床研究中,受试者每疗程使用palifosfamide(初始剂量为100 mg·m-2·d-1,d1 ~ d3)和多柔比星(初始剂量为60 mg·m-2·d-1,d1),1 个疗程为 21 天,上述两药剂量随疗程个数的增加逐渐升至各自的MTD,即分别为150和75 mg·m-2·d-1。73 个疗程后的治疗结果显示,该联用方案可被较好地耐受,未见DLT,未出现脑部病变、出血性膀胱炎及肾脏毒性。与本研究相关的3/4级不良反应包括中性粒细胞减少和血小板减少。在12名研究结果可供评价的受试者中,3名病情部分缓解,无进展生存期(PFS)中位数为20周。
研究人员在患有手术无法切除或转移性软组织肉瘤的受试者中开展了一项名为PICASSO、多中心、平行、随机的Ⅱ期研究,以比较palifosfamide+多柔比星联用方案与单用多柔比星的疗效。研究的主要评价指标为PFS,次要指标为治疗方案的安全性、耐受性和总体应答率。共有20名受试者发生病情恶化,其中14名单独使用多柔比星,6名接受联合治疗方案。结果显示,联用组受试者的PFS为7.8个月,而多柔比星组仅为4.4个月,风险比为0.427 (P=0.019);联用组和多柔比星组的缓解率分别为23%和9%。一项安全性数据的中期分析报告指出,palifosfamide不会增加多柔比星的毒性。两组中最常见不良反应为中性粒细胞减少和疲劳,其次为低钾血、恶心、贫血、白细胞减少和脱发。
ZIOPHARM Oncology公司现已启动了一项名为PICASSO3、多中心、随机、双盲、安慰剂对照的关键性Ⅲ期临床试验,共有424名未曾接受化疗的转移性组织肉瘤患者参加,旨在分析比较palifosfamide+多柔比星与多柔比星+安慰剂的安全性和有效性,以供该药提交审评时的需要。其中加速审批的主要考察指标为PFS,完全审批的主要考察指标为总生存率。该研究在位于北美、欧洲、南非、澳大利亚、以色列和韩国约150个中心进行。此外,ZIOPHARM Oncology还计划扩大palifosfamide的适应证,如儿科癌症、淋巴瘤和乳腺癌等。
R 979.1
(杨臻峥 编译)
