灭菌注射用水分装及灭菌生产工艺验证
2011-01-31蒋井明谈岩舫卫天萍
席 亮 蒋井明 谈岩舫 高 军 卫天萍
根据2010年版《药品生产质量管理规范》附录1“无菌药品”第61条的要求,无菌药品应尽可能采用加热方式进行最终灭菌,最终灭菌产品中的微生物存活概率(即无菌保证水平,SAL)不得高于10-6。良好的无菌药品生产工艺是保证药品安全性最可靠的手段,采用湿热灭菌方法进行最终灭菌的,通常标准灭菌时间F0值应当大于8 min。因此,通过对灭菌注射用水分装、灭菌生产工艺的质控参数、质控标准的检查确认,以积累的生产、检验和其他历史数据为依据,通过统计学分析,证明所采用的分装、灭菌生产工艺能够满足产品安全性、生产可靠性的要求。
灭菌注射用水(Sterile Water for Injection),本品为无色的澄明液体、无臭、无味,每支0.5 ml(实际分装量0.7 ml/支),主要成分∶注射用水。按照注射剂生产工艺制备,用于冻干疫苗的稀释剂。
1 生产工艺
1.1 生产工艺描述
安瓿分装的灭菌注射用水为最终灭菌产品,整个工艺按照生产顺序分为配制、洗瓶、干热灭菌、灌封、检漏、高压灭菌。由不同的生产人员在不同洁净级别的房间内操作,D级房间进行灭菌注射用水的安瓿清洗、干热灭菌,C级房间局部A级进行配制和安瓿的灌装、封口。
1.2 分装、灭菌生产工艺流程图(如图1所示)
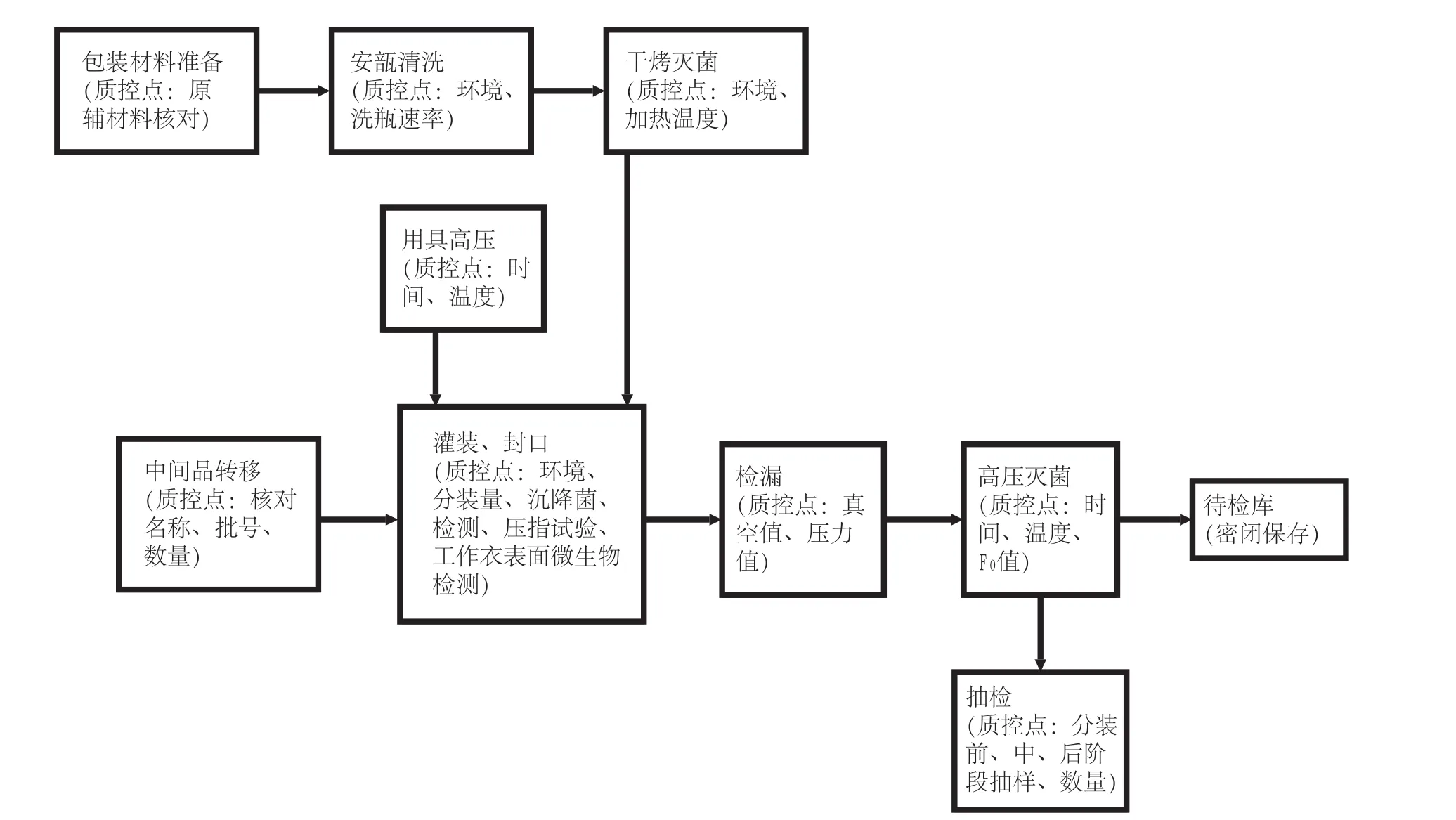
图1 分装、灭菌生产工艺流程图
2 技术和支持文件的检查确认
2.1 检查生产工艺文件(见表1)

表1 生产工艺文件
2.2 检查原材料质量标准
生产前内包装材料必须通过QC按照确定的检测方法和质控标准检定,证实合格后方能投入生产(见表2)。

表2 成品和原材料标准
2.3 检查确认检定方法验证文件(见表3)

表3 检定方法验证文件
2.4 检查确认主要仪器设备情况(见表4)
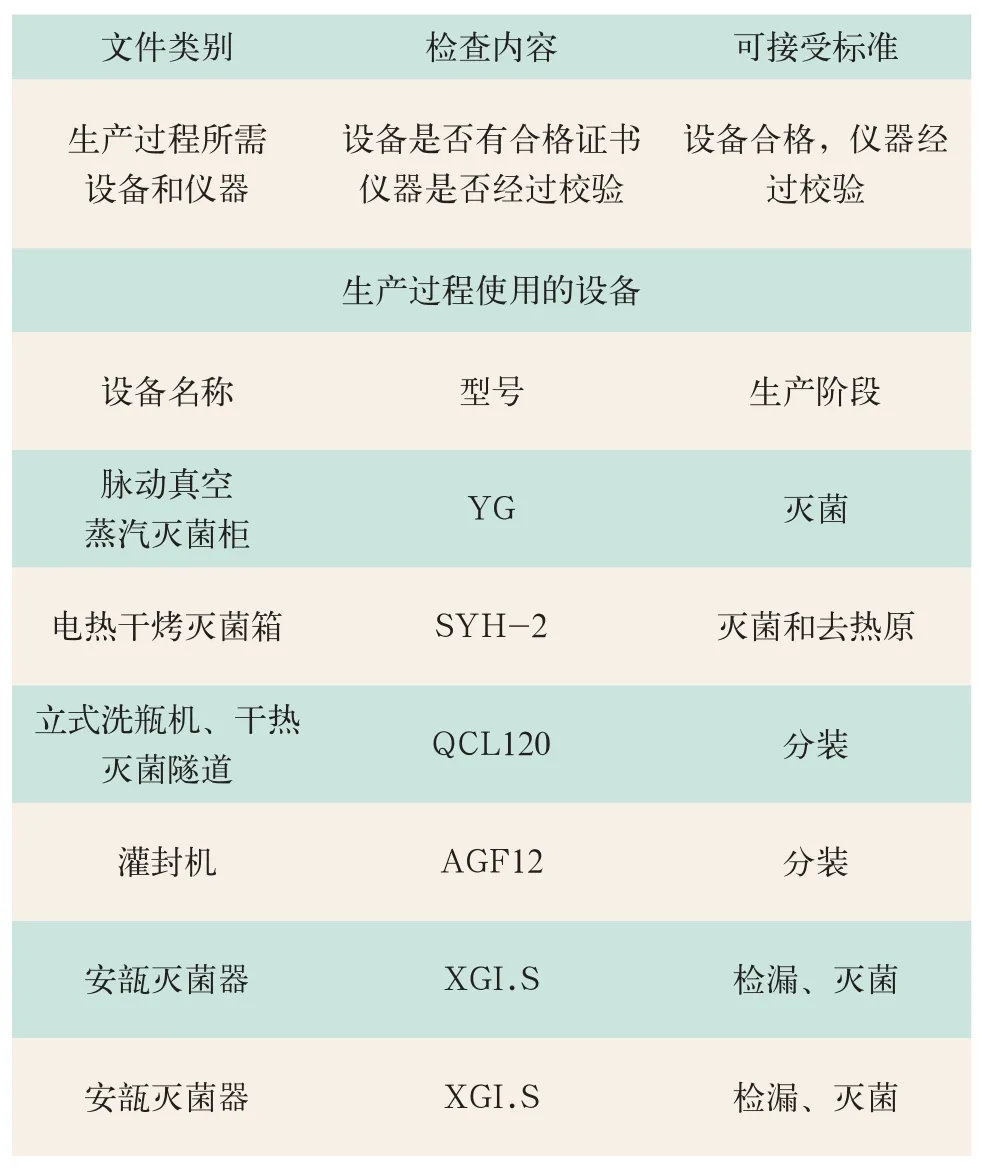
表4 主要仪器设备情况
2.5 检查确认记录
填写检查确认记录。如有偏差,在偏差记录中记录验证过程中所发生的偏差。
3 生产过程的验证
3.1 分装过程的验证(见表5)
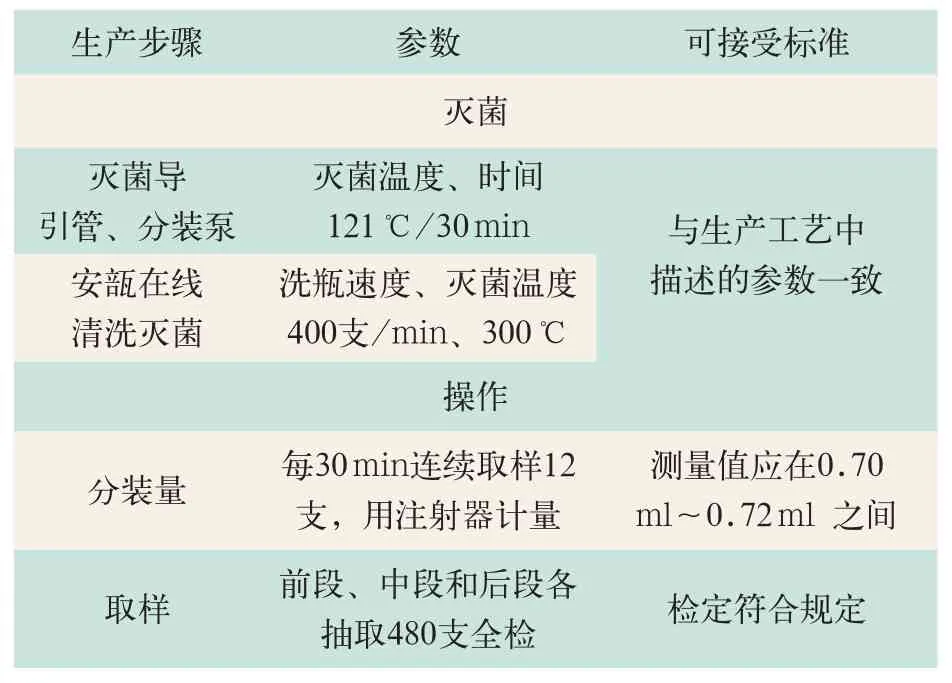
表5 检查确认分装过程关键参数
3.2 检查确认记录
填写分装过程的确认记录。如有偏差,在偏差记录中记录验证过程中所发生的偏差。
3.3 灭菌过程的验证(见表6)

表6 检查确认灭菌过程关键参数
3.4 检查确认记录
填写灭菌过程的确认记录。如有偏差,在偏差记录中记录验证过程中所发生的偏差。
3.5 生产环境、设施和设备的确认
检查确认洁净区环境参数(温度、压力、污染等),检查确认电热干烤灭菌箱、干热灭菌隧道器、脉动真空蒸汽灭菌柜和安瓿灭菌器热穿透试验(见表7)。

表7 检查确认生产环境、设施和设备的关键参数
3.6 检查确认记录
填写生产环境、设施和设备的确认记录。如有偏差,在偏差记录中记录验证过程中所发生的偏差。
4 统计学分析
统计2011年生产的灭菌注射用水64批共计138亚批的F0值,统计学分析如下
4.1 最大值、最小值、算数平均值
max=11.10 min=8.80 X=9.64
4.2 标准差 (SD)、安全上限、安全下限
SD=0.42 X+3×SD=10.9 X-3×SD=8.4
4.3 2011年灭菌注射用水高压灭菌F0值趋势图(如图2所示)

图2 2010年灭菌注射用水高压灭菌F0值趋势图
4.4 数据结果
统计2011年生产的灭菌注射用水共138亚批的F0值,其中135个亚批的F0值在安全上限、安全下限之间;另外,有3个亚批的F0值超过安全上限,F0值分别为11.1、11.0、11.0,在偏差记录中记录所发生的偏差,对所发生的偏差进行调查。
5 偏差处理
5.1 在偏差目录中记录在验证过程中所发生的所有偏差(见表8)

表8 偏差记录
5.2 可接受标准
偏差调查∶对出现的每一个偏差按照偏差控制程序进行调查,处理每个偏差并完成偏差报告,所有偏差都应得到纠正和解决。(见表9)
5.3 检查确认记录、填写偏差记录
6 验证周期
每年1次或者在下列情况需要及时验证∶关键设备大修或更换;工艺、方法、主要原材料有变更。
7 可接受标准
需达到以下要求∶必须符合方案所述的要求,最终产品达到成品检定标准。

表9 偏差调查处理记录
[1]国家药典委员会.中华人民共和国药典三部[M].2010年版.北京∶中国医药科技出版社.2010.
[2]国家食品药品监督管理局.药品生产质量管理规范(修订版)[S].北京∶国家食品药品监督管理局,2010.
[3]国家食品药品监督管理局药品安全监管司、药品认证管理中心.药品生产验证指南[M].北京∶化学工业出版社,2003.
[4]李钧.药品GMP验证教程[M].北京∶中国医药科技出版社,2003.
[5]国家食品药品监督管理局药品认证管理中心.欧盟药品GMP指南[M].北京∶中国医药科技出版社,2008.
