与神经系统相关的儿童线粒体疾病
2011-01-22TonDegrauw,迟景上,王艺

TonDegrauw 教授迟景上 教授王 艺 教授
王 艺教授 欢迎Ton教授和迟景上教授参加2010年上海神经精神国际论坛,并受邀参加由《中国循证儿科杂志》编辑部组织的线粒体疾病的现状专家对谈录。随着检测技术的发展,能量代谢性疾病越来越引起内科医生的重视,尤其是线粒体疾病,由于多数累及中枢神经系统,但其表现型多样,临床认识非常有限,请Ton教授结合您在2010年上海神经精神国际论坛上的这方面专题报告中的系列疾病做一介绍。

迟景上教授 当前国际上是否有对线粒体疾病进行新的分类?
Ton教授 相关组织试图根据分子学基础进行新的分类,目前基因研究小组对线粒体基因进行增殖融合,对电子传递链酶复合物Ⅰ~Ⅴ的蛋白结构进行分类。可能很快就有新的线粒体疾病分类。
王 艺教授 目前,在诊断线粒体疾病方面还有很大的困难,请两位教授介绍一下你们的经验。
Ton教授 在辛辛那提儿童医院诊断为线粒体病的患儿中,只有20%的患儿具有临床综合征的表现,依靠临床表现和神经系统检查,神经科医生可以做出诊断,但是80%患儿诊断比较困难。当遇到这部分患儿时,首先进行血液和尿液的遗传代谢筛查,包括葡萄糖、丙酮酸、乳酸、丙氨酸、肌酸、胍基乙酸、β-羟丁酸、肉碱和酰基肉碱等,对于ETC缺陷的患儿来说,筛查是最基础的检查。在线粒体疾病患儿中有60%~70%所有的筛查结果可能都是正常的。
其次,可考虑行脑部MR和质子磁共振波谱(MRS)检查。由于N-乙酰天冬氨酸(NAA)峰在3.0 ppm,谷氨酸邻近NAA,在3.1 ppm左右,甘氨酸在3.5 ppm左右,所以3.0 tesla波谱检查更容易区分这些代谢峰。当然,我们正在进行4.0 tesla场强的MR研究,相信有些大学还有5.0 tesla场强的检查。
如果通过上述检查仍不能排除线粒体疾病,则应进行线粒体DNA检测,不过线粒体DNA突变的发生率很低,仅占30%左右。尤其是核型的检测,很难发现异常。
肌肉活检组织化学染色,电镜观察线粒体的结构形态,ETC酶复合物Ⅰ~Ⅴ活性的检测。目前有2种方法可使用冰冻肌肉组织或者新鲜的肌肉组织分离线粒体检测这些复合物的活性,低于25%为异常。但是对于哪一种检查方法获得的结果更好目前仍然存在争议。线粒体疾病约50%存在ETC酶复合物缺陷。Leigh病可出现复合物Ⅳ的缺陷。美国辛辛那提儿童医院1年进行100例肌肉活检并进行ETC活性的检测。
线粒体病患儿的症状往往看似不是神经系统疾病,我就曾遇到过1例11岁的男性患儿,4年来出现渐进性乏力,主诉最多能行走300米,体格检查和神经系统查体均正常,学习成绩优秀,既往体格健康。心电图和肌电图均正常。但值得一提的是,该患儿就诊时身高仅处于P40水平,但在5年前还处于P90水平。代谢检查:葡萄糖74 mg·dL-1,乳酸5.5 mmol·L-1,丙酮酸0.10 mmol·L-1,乳酸/丙酮酸为55,丙氨酸 801 μmol·L-1(正常值:195~499μmol·L-1),辅酶Q10正常,尿有机酸检测示乳酸和β-羟基丁酸水平均增高。
该患儿尚进行了ETC活性测定,复合物Ⅰ和Ⅲ的活性降低(10.8%),复合物Ⅳ的活性仅为15.9%,复合物Ⅲ的活性为41.1%,故考虑该患儿为复合物Ⅰ和Ⅳ缺陷。
电镜检查结果可见该患儿体内存在有趣的线粒体结构,圆形的线粒体沿着胞膜以环状排列(图1A),在其他位置还有以方形结构存在的线粒体(图1B)。
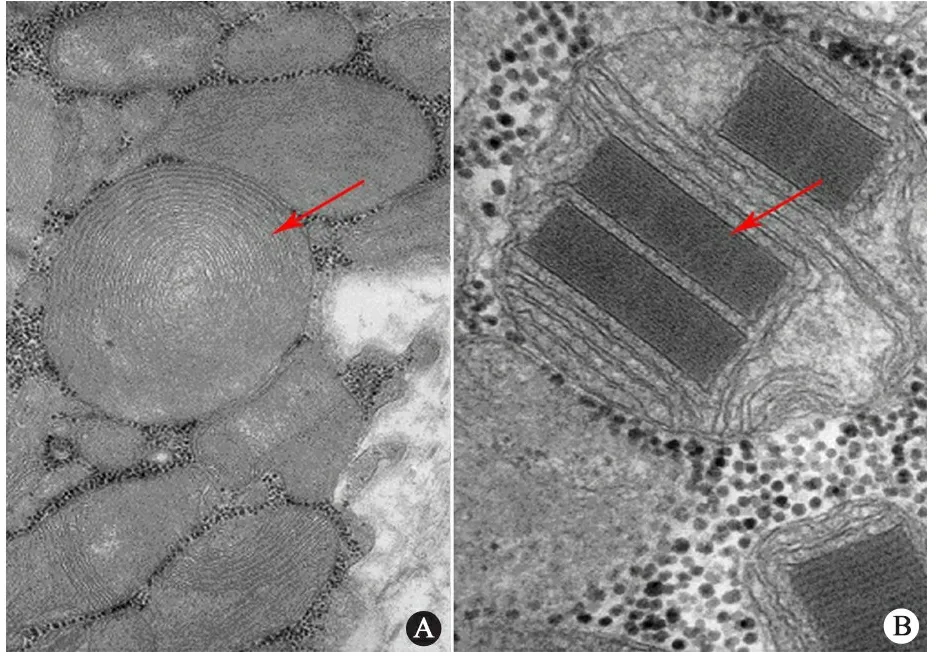
图1 线粒体病患儿电镜检查结果所见
注 A:红色箭头所示可见圆形环状排列的线粒体结构;B:红色箭头所示可见方形线粒体结构
同时亦对该患儿进行了线粒体DNA完整基因组测序,结果发现该患儿存在m.5590G>A (tRNA Ala) 同质异常变异,该变异位于高度保守的氨基酸连接部位(tRNA Ala),由原本的G-C 配对突变为G-U错配。曾有报道单纯性肌病患者(症状与本病患儿类似)合并复合体Ⅰ和Ⅳ缺陷者伴有m.5591G>A(邻近位点)突变,但其母亲无该位点突变。
本例患儿可能是以骨骼肌受累为主的一种新的线粒体疾病。目前尚无针对该患儿的特异的治疗手段,接下来唯一能做的就是对该患儿进行长期随访,观察其心脏、眼、耳朵、是否发生糖尿病和甲状腺功能等。本病例为典型的临床上看似不是神经系统疾病,但患儿所表现出的症状往往应引起临床医生的高度警惕,要考虑到线粒体病的可能性。
王 艺教授 迟教授,中国台湾地区目前的检测手段如何?
迟景上教授 在中国台湾地区也会遇到一些代谢性疾病的患儿,但是由于文化背景的不同,在中国台湾很难说服家长进行肌肉活检,与美国辛辛那提儿童医院的诊断步骤还是有所不同,当疑似神经代谢疾病的患儿,除了血及尿液的代谢物检测外,进行MR、MRS和口服葡萄糖乳酸测试的检查,如果影像学检查和(或)口服葡萄糖乳酸测试异常就进行分类,区分时是综合征型还是非综合征型?如果是综合征型的,则进行DNA序列检测,检测结果正常,就说服家长进行肌肉活检。
在中国台湾地区目前较少有肌肉活检后的生物化学酵素检查,进行肌肉细胞染色及电镜检测,如果电镜中存在结构异常诊断线粒体病。如果电镜检查正常则随访,且保留患儿的血样以用于特异性基因的检查。
口服葡萄糖乳酸的测试的方法是:患儿禁食8 h,早上8点检测基础乳酸,如果患儿不合作先给予镇静,然后给予1.75 g·kg-1的葡萄糖口服液,在随后的3 h中,每30 min检测1次乳酸和丙酮酸,如果60和90 min的乳酸水平升高大于基础量的5 mg·L-1,则表示有异常。
王 艺教授 目前对线粒体疾病的分子基础有哪些新的认识?有相关的基因学检测技术吗?
Ton教授 线粒体疾病的分子学基础与线粒体DNA突变有关,目前认为KSS综合征、Pearson综合征、散发的进行性眼外肌瘫痪(Sporadic PEO)、糖尿病伴耳聋等与mRNA重排有关系。与结构蛋白编码基因突变有关的有LHON、神经性肌无力、共济失调伴视网膜色素变性(NARP)和母系遗传性Leigh′s综合征。与tRNAs编码基因有关的有MELAS综合征、MERRF综合征、母系遗传成人发病的合并心脏病变的线粒体肌病(MIMyCa)、进行性眼外肌瘫痪(PEO)、单纯性肌病、糖尿病伴耳聋、感音神经性耳聋、肥厚型心肌病和脉管病。氨基糖苷类抗生素诱导性耳聋和肥厚型心肌病与rRNAs编码基因有关。当然上述的多数仅适用于基础研究。
如果患儿高度怀疑Leigh综合征,应进行SURF1基因检查。Leigh′s综合征患儿可出现电子传递链复合物Ⅳ的缺陷,所以对检测到复合物IV缺陷的患儿要进行SURF1基因检查。但是基因检查比较昂贵,因此检查必须具有针对性。
迟景上教授 目前中国台湾地区只能进行SURF1和2的检测,不能进行POLG基因的检测。
王 艺教授 复旦大学附属儿科医院现有的辅助诊断方法也有限,可以做的是血和尿液的遗传代谢筛查,影像学方面可以进行MR和MRS的检查,没有针对线粒体疾病的患儿开展肌肉活检,也没有相关基因学检查的手段。
迟景上教授 线粒体量的缺失综合征的情况如何呢?
Ton教授 线粒体量的缺失综合征也并不十分罕见,临床表现型和线粒体功能结构变化所致的表现型相仿。肌肉活检往往发现ETC酶复合物减低,Ⅰ,Ⅱ,Ⅲ,Ⅳ可以很低,当检测到多重的复合物降低时,要考虑线粒体量的缺失综合征。需要进行线粒体DNA的定量检测,会显示分子数量是否正常。如果发现分子数量很低的话,会进一步进行TK2基因的核型检测。
另外,对辅酶Q10缺失综合征非常感兴趣,该综合征可表现为单纯的肌病,进行性进展,多数伴随肌无力,也可伴随脑病和肝脏问题,也可表现为肌肉、神经和脑混合型。当进行肌肉ETC活性检测时,同时检测Ⅰ和Ⅲ及Ⅱ和Ⅲ时,会发现复合物Ⅰ和Ⅲ活性减低,Ⅱ和Ⅲ活性减低,但单独进行各复合物检测时均为正常。因为辅酶Q10连接复合物Ⅰ、Ⅲ以及复合物Ⅱ、Ⅲ。美国辛辛那提儿童医院正在发展检测肌肉辅酶Q10的技术。辅酶Q10对这一疾病的治疗效果很好。目前为止对辅酶Q10的研究已有2年了。
王 艺教授 目前对线粒体疾病的治疗方面有没有新的进展?
Ton教授 对每个医生来说,最尴尬的事情就是缺乏有效的治疗,这一点可能会阻碍线粒体疾病研究的进一步进展。但是对于肌酸代谢障碍中的合成障碍,目前明确高剂量的肌酸具有很好的替代治疗效果,该药物即将进入临床试验阶段。
王 艺教授 很荣幸能请到2位教授,让我们对线粒体疾病的现状有了更深入的了解,希望今后有机会继续交流。
