具抗病毒活性的2',3'-双脱氧核苷合成策略
2011-01-12刘彩连黄海燕姚其正
张 敏, 刘彩连, 李 战, 黄海燕, 姚其正*
(1.中国药科大学药学院,江苏南京210009;2.南京长澳医药科技有限公司,江苏南京210008)
2',3'-双脱氧核苷(ddNs)是一种结构特殊的核苷类似物,能通过以下3个主要途径产生抗病毒活性:1)抑制病毒核酸复制所需酶的活性;2)阻抑病毒中参与翻译和帽化mRNA以及插入RNA的酶的活性;3)直接阻断病毒DNA的合成[1]。在这些抗病毒的化学小分子中,有些已作为抗艾滋病毒(HIV)和/或抗乙肝病毒(HBV)的药物用于临床[2-5],如齐多夫定(AZT)、地丹诺辛(dd I)、扎西他宾(ddC)、司他夫定(d4T)、拉美夫定(3TC)、阿巴卡韦(ABC)和洛德腺苷等,其共同的结构特征是:与氮杂环碱基(嘧啶或嘌呤)相连的戊(杂)环糖的2'和3'位上无羟基。按结构它们还可进一步分为4小类:1)2'和3'位有其他基团取代的ddNs,如AZT和洛德腺苷;2)2'和3'位无取代的ddNs,如dd I和ddC;3)2',3'-双脱氧-2',3'-双脱氢核苷(d4Ns),如d4T;4)其他类型的ddNs,如3TC和ABC。在这4小类ddNs中,第2类ddNs被研究和应用得最多,其合成方法的研究一直受到药物化学家的关注。
ddNs的合成通常有两条途径:一是先合成各种2,3-双脱氧糖类衍生物,然后再将其分别与各种氮杂碱基经糖苷化反应制得ddNs;二是分别以2'-脱氧核苷和核苷为原料,通过糖环上的各种选择性脱氧反应制得ddNs。由于2,3-双脱氧糖类的制备过程较复杂以及糖苷化副反应较多等原因,用第一条途径合成ddNs的方法研究现已很少,而近10年来,第二条途径的合成方法研究发展迅速,报道较多,本文对此作一综述。
以2'-脱氧核苷(1)或核苷(2)为原料制备ddNs有两种策略:其一,将原料经多步反应,直接制得ddNs,即路径1;其二,先将原料经反应制得重要中间体d4Ns,然后再将其还原制得ddNs,即路径2(见图1)。

图1 以2'-脱氧核苷或核苷为原料合成ddNs的两种策略Figure 1 Two strategies for ddNs synthesiswith 2'-deoxynucleosides or nucleosides as startingmaterials
1 以2'-脱氧核苷为原料制备2',3'-双脱氧核苷
1.1 经3'-脱氧反应的路径1合成策略
用2'-脱氧核苷制备ddNs的方法最早由Todd等(JChem Soc,1955年)提出,该法仅需将2'-脱氧核苷中5'-OH进行选择性保护后,经3'-脱氧反应与脱保护,即可得ddNs(见图2)。该法已成为合成ddNs的主要方法之一,其关键是:在3'-脱氧反应中,必须将3'-OH置换为各种易于脱去的基团,如用碘原子、甲磺酰氧基(MsO)、对甲苯磺酰氧基(TsO)和乙硫基(EtS)等,这些置换的基团都可在金属催化剂(如Pd-BaSO4、Pd-C或Raney Ni等)作用下被加氢并消除,从而制得ddNs[6]。
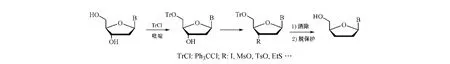
图2 经3'-脱氧反应合成ddNsFigure 2 Synthesis of ddNs via 3'-deoxygenation
1.2 路径2合成策略
“1.1”节的方法适用于经嘧啶类和嘌呤类2'-脱氧核苷制备ddNs,但对于嘧啶类ddNs的合成,还可利用嘧啶类2'-脱氧核苷的结构特点,通过中间体d4Ns的还原而实现,如2',3'-双脱氧胸苷(ddT,3)的合成(见图3)。
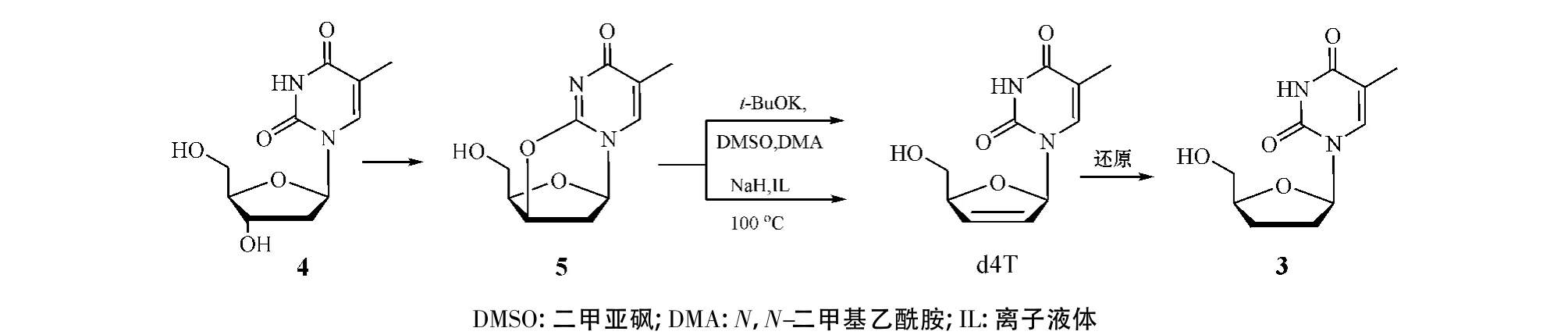
图3 经中间体d4T合成ddTFigure 3 Synthesis of ddT via intermediate d4T
由图3可见,将原料β-胸苷(4)中嘧啶环与糖环经脱水制得2,3'-脱水胸苷(5),以往常用t-BuOK为碱试剂,以DMSO或DMA作溶剂,对化合物5进行碱催化的β-消除,即可制得2',3'-不饱和核苷d4T[7]。然而,其中消除反应(5→d4T)常常不能令人满意,如反应时间长、溶剂DMSO或DMA沸点高、分离难、收率低等。近年,Kumar等[8]采用咪唑类IL(如[MoeMIm][Ms]、[MoeMIm][TFA]和[BMIm][TFA])作溶剂、NaH作碱试剂,对2,3'-脱水胸苷进行碱催化消除反应,使反应时间缩短到5~10 min,分离方便,d4T的产率提高至90%左右(见表1);最后,再将d4T还原成ddT。这是对被称为绿色溶剂的咪唑类IL用于核苷衍生物合成的首次报道,极具研究与应用价值。

表1 不同条件下碱催化消除反应的时间和收率Table 1 Timeandyieldofeliminationreactioncatalyzedbybaseunderdifferentconditions
2 以核苷为原料制备2',3'-双脱氧核苷
以核苷为原料制备ddNs时,需要脱除糖环上2'和3'位上的两个羟基,因而,研究人员常采用路径2经中间体d4Ns的合成策略,且近年来相关合成方法研究也取得了长足进展。
2.1 路径2经中间体d4Ns的合成策略
2.1.1 利用Mattock反应的合成方法
Mattock反应是由Mattock等(JChemSoc,1964年)创立而得名,即将试剂α-乙酰氧基异丁酰溴或α-乙酰氧基异丁酰氯(α-AIBBr/Cl,也称Mattock试剂,6)分别与苄醇和乙二醇进行的反应(见图4)。
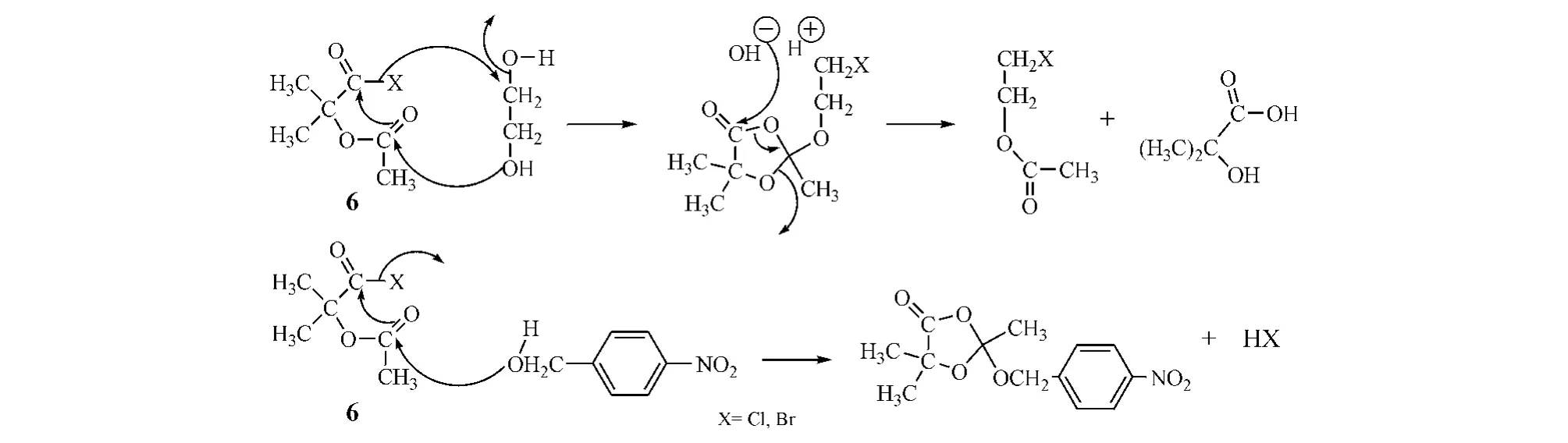
图4 Mattock试剂与羟基化合物的反应机制Figure4 ReactiveprocessofMattockreagentwithhydroxylcompounds
由图4可见,α-AIBBr/Cl与单伯醇和邻二醇反应分别有着不同机制。受Mattock反应机制的启发,Moffatt等(JAmChemSoc,1973)首先将Mattock试剂引入到合成嘌呤类2',3'-双脱氧腺苷(ddA)中,此试剂与核苷反应,即得到高产率的5'位为1,3-二烷-4-酮-2,5,5-三甲基环状取代基而2'位卤化及3'位乙酰化的产物(7)和2'位乙酰化及3'位卤化的产物(8)的混合物,副产物较少;然后经Zn或Zn-Cu还原消除脱去2'和3'位上卤化酰化的基团,形成中间体d4Ns,最后用Pd-C催化加氢还原,得到ddNs(见图5)[9]。
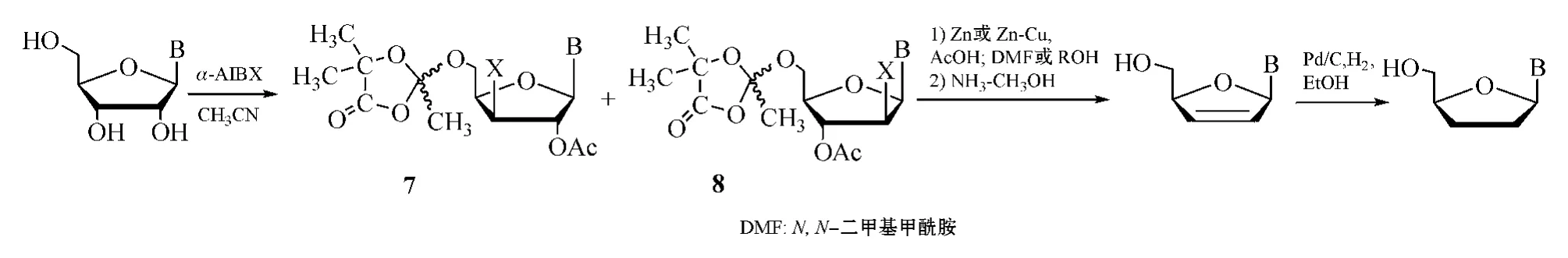
图5 核苷经Mattock反应合成ddNsFigure5 SynthesisofddNsviaMattockreaction
值得注意的是:核苷经Mattock反应在2'和3'位上形成的取代基互为反式共平面,有利于随后的还原消除反应,此方法已成为由嘌呤类核苷制备d4Ns和ddNs的一条重要途径。
该反应同样适用于以嘧啶类核苷为原料合成ddNs,只是嘧啶类核苷[如尿苷(9)]与Mattock试剂反应时,仅得到2'-卤化物,且5'位的取代反应受溶剂影响较大(见图6)[10],然而这些并不影响随后d4Ns和ddNs的合成。

图6 M attock试剂与尿苷在不同溶剂中的反应Figure 6 Reaction of Mattock reagentwith uridine in different solvents
虽然Mattock反应可用于合成ddNs,但由于Mattock试剂存在易吸湿、稳定性差、价格贵等问题,使其应用受到较大限制。
2.1.2 利用其他酰卤反应的合成方法
由于嘧啶分子的结构不同于嘌呤,以嘧啶类核苷为原料制备ddNs时,可用价廉、来源广泛的乙酰溴等酰卤代替高价的Mattock试剂来制备酰化卤化中间体。例如,将尿苷在乙腈中与乙酰溴反应可得收率达80%的酰化卤化产物[11],但在随后用Zn等进行还原消除反应制备d4N时,易发生糖苷键断裂的副反应(见图7)[12],其原因可能是:嘧啶类核苷与一般酰卤作用时,首先形成一个关键中间体——2,2'-脱水嘧啶核苷,卤代后,使2'位卤素和3'位酰化基团处于顺式构型,而2'位卤素与1'位C-N糖苷键却呈反式共平面[13],从而导致还原消除反应中糖苷键易断裂,影响d4Ns及ddNs的收率。
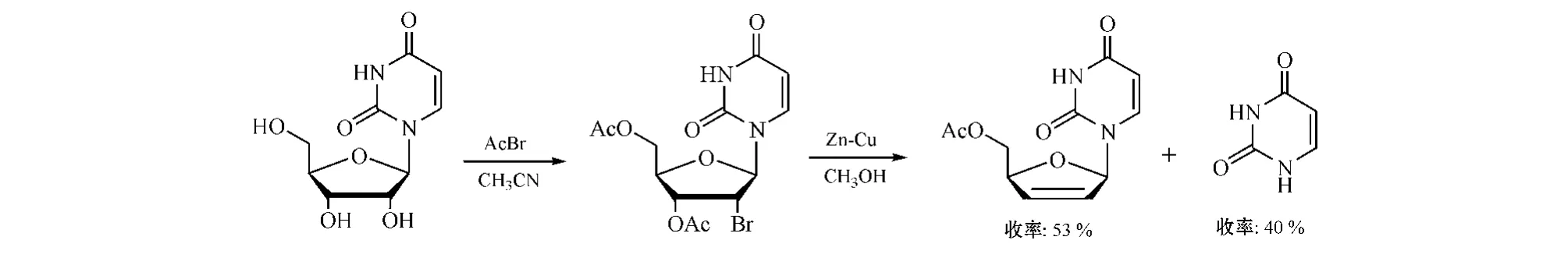
图7 尿苷与乙酰溴反应及随后的还原消除反应Figure 7 Reductive elimination reaction following reaction of uridine and acetyl bromide
为克服上述问题,研究者们采用以下各种手段对此合成方法进行了改进:
1 )原乙(或甲)酸三酯法。为了使核苷2'和3'位上的卤素与酰氧取代基呈互为反式共平面,有人先将核苷[如5-甲基尿苷(10)]在酸作用下与原乙酸三甲酯反应,致糖环2'和3'位上邻二羟基转化成环醚结构,再与酰卤反应,取代2',3'-环醚,形成2'和3'位上互为反式共平面的卤代和酰化的中间体混合物,用Zn还原消除时,很好地避免了C-N糖苷键断裂的副反应,保证了d4Ns和ddNs的收率(见图8)[12,14]。此法也适用于以嘌呤类核苷为原料合成ddNs[15]。
原甲酸三甲酯亦可代替原乙酸三甲酯用于此法,不过通常以对甲苯磺酸作催化剂[16-17],所得2',3'-环醚衍生物再在乙酐中高温回流,直接转化为d4N衍生物,这样虽减少了反应步骤,但高温回流反应的收率并不高(见图9)[17]。
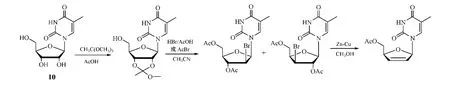
图8 原乙酸三甲酯参与的d4Ns合成反应Figure 8 Synthesis of d4Ns using trimethyl orthoacetate
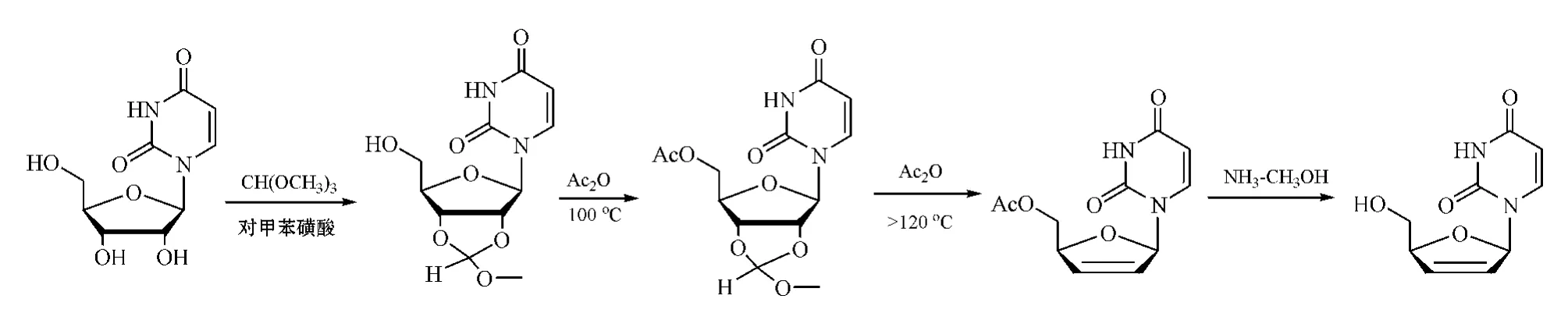
图9 原甲酸三甲酯参与的d4Ns合成反应Figure 9 Synthesis of d4Ns using trimethyl orthoformate
相对于Mattock反应,利用原甲(或乙)酸三甲酯制得2',3'-环醚中间体、再经 d4Ns还原合成ddNs的方法,较为合理、经济,所用原料价廉易得,现已发展成为一种较大规模生产d4Ns及ddNs的方法。
2 )核苷2',3'-环氧化法。Chen和Cos-tenaro等[18-19]以5-甲基尿苷为原料,设计了另一种d4Ns和ddNs合成方法:将制得的2',3',5'-三-O-甲磺酰基-5-甲基尿苷用1 mol·L-1氢氧化钠水溶液处理,经脱水得到5'-O-甲磺酰基-2',3'-环氧-5-甲基尿苷(11),再用乙酰溴/甲醇将2',3'-环氧打开,生成得3'-溴代-5-甲基阿糖尿苷衍生物和2'-溴代-5-甲基木糖尿苷衍生物的混合物,并用乙酰溴/二氯甲烷酰化,所得2'与3'位取代基处于反式共平面状态,经Zu/Cu还原消除得到高收率的 d4T衍生物(见图10),然后在碱性条件下脱去5'位上甲磺酰基,Pd-C催化加氢还原,最终得到ddNs。此法也适用于以嘌呤类核苷为原料合成ddNs。

图10 利用核苷2',3'-环氧化法合成d4NsFigure 10 Synthesis of d4Ns by epoxidation of nucleoside
3 )核苷 3'-甲磺酰化法。Chen和靳立人等[20-21]利用甲磺酰化法先将尿苷衍生物转化成2',3',5'-O-三甲磺酰基-(5-甲基)尿苷,再在强碱条件下经脱水得到2,2'-环氧-尿苷中间体,然后经酰化、溴化生成2'-溴-3'-O-甲磺酰基-5'-苯甲酰氧基尿苷,而由于甲磺酰基是一很好的离去基团,即使2'与3'位取代基处于顺式状态,但在锌粉作用下,也可几乎定量地还原消除得到d4T/d4U衍生物,再经脱保护基即得d4T/d4U,不会发生C-N苷键断开现象(见图11)。这条合成路线虽较长,但总收率较高。
此外,2'-溴-3'-O-甲磺酰基-5'-苯甲酰氧基尿苷亦可在偏碱性条件下,应用Lindlar催化剂(Pd-BaSO4)催化,直接进行加氢还原,得到收率较高的d4T/d4U(见图11)[22]。
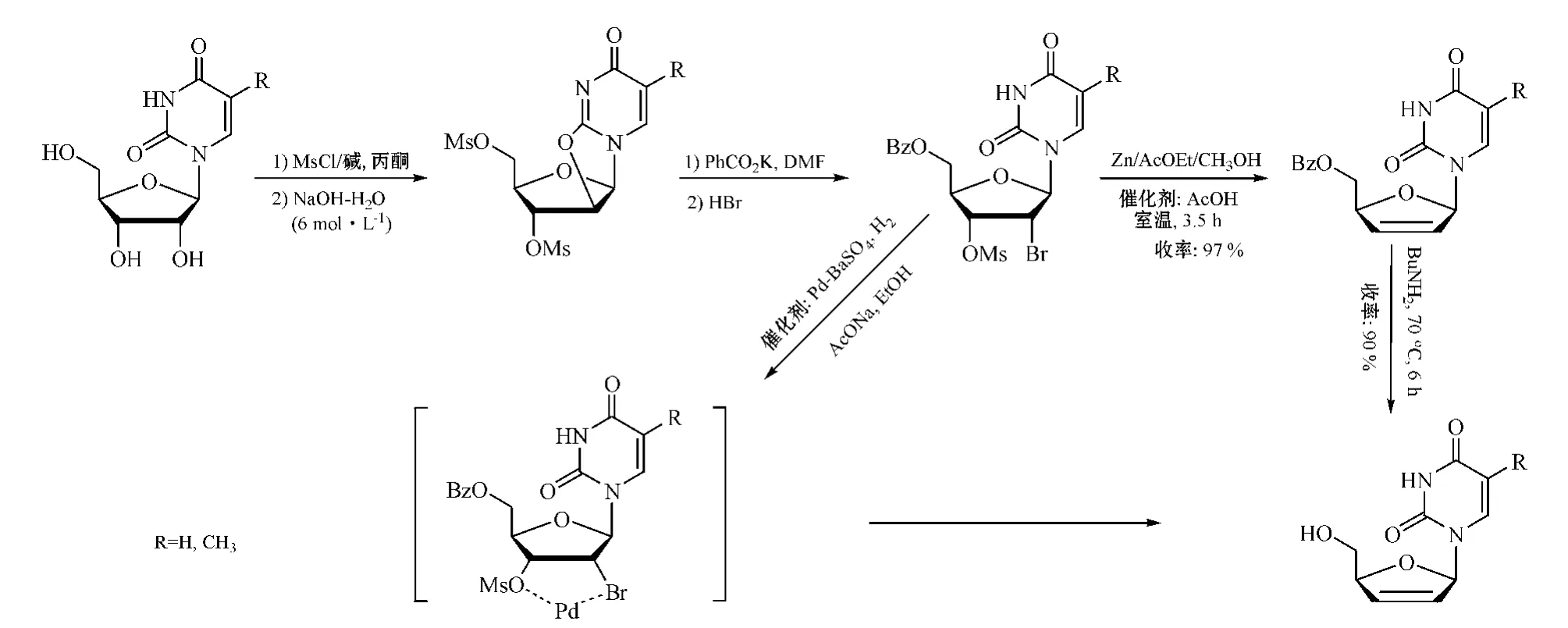
图11 利用核苷3'甲磺酰化法合成d4NsFigure 11 Synthesis of d4Ns by 3'-methanesulfonation of nucleoside
2.1.3 利用Barton-McCombie自由基脱氧反应的合成方法
Chun等[23]参照Nair等(J Am Chem Soc,1989年)曾提出的方法(即Nair法),用1,1'-硫代羰基二咪唑(Im2C S)和核苷反应,使糖环的2',3'-二仲羟基转变为2',3'-环硫代碳酸二酯(12),再经Barton-McCombie自由基脱氧反应[即利用自由基引发剂2,2'-偶氮双异丁腈(AIBN)和链载体三丁基锡化氢(n-Bu3SnH)所组成的试剂进行的反应(Barton,J Chem Soc Perkin Tran 1,1975年)],得到两种中间体(13和14);然后用Et4NF/CH3CN与中间体13反应,得d4Ns,最后制得ddNs;而中间体14则再经Im2C S处理,得3'-咪唑硫代甲酸酯衍生物,最后用Et4NF消除处理,合成ddNs(见图12)。
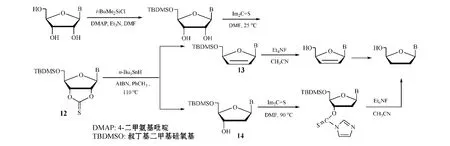
图12 经Barton-M cCombie自由基脱氧反应合成d4Ns和ddNsFigure 12 Synthesis of d4Ns and ddNs via Barton-McCombie radical deoxygenation reaction
Torii等[24]用咪唑、二硫化碳和碘甲烷代替Im2C S与核苷反应,得到核苷的2',3'-二-O-硫代碳酸酯衍生物,再用n-Bu3SnH和AIBN进行脱氧反应,即得ddNs的前身d4Ns(见图13)。这是改进的Nair方法,又称改进的Barton-McCombie自由基脱氧反应。
化合物12也可用硫代光气(Cl2C S)制得,且因Barton-McCombie自由基脱氧反应中所用链载体三丁基锡化氢毒性较大,现已改用亚磷酸三甲(或三乙)酯(即经Corey-Winter烯化反应)或者1,3-二甲基-2-苯基-1,3,2-二氮磷杂环戊烷等膦化物来处理化合物12,再经脱氧得到d4Ns[25]。其机制即通过磷夺去硫代羰基上的硫,形成不稳定的中间体,再经分解释放出CO2,达到脱氧之目标(见图14)。
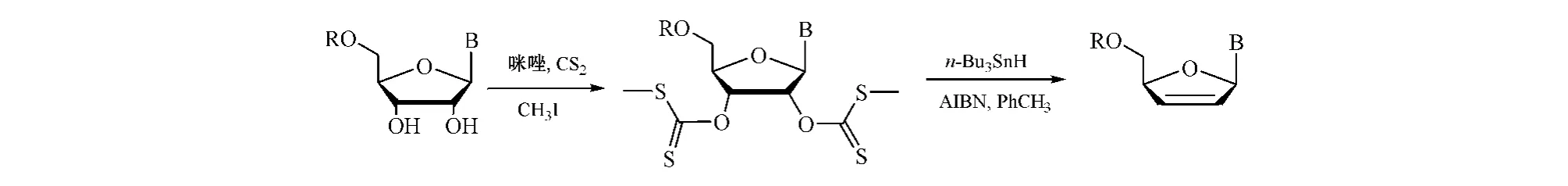
图13 经改进的Barton-M cCombie自由基脱氧反应合成d4NsFigure 13 Synthesis of d4Ns via the improved Barton-McCombie radical deoxygenation reaction
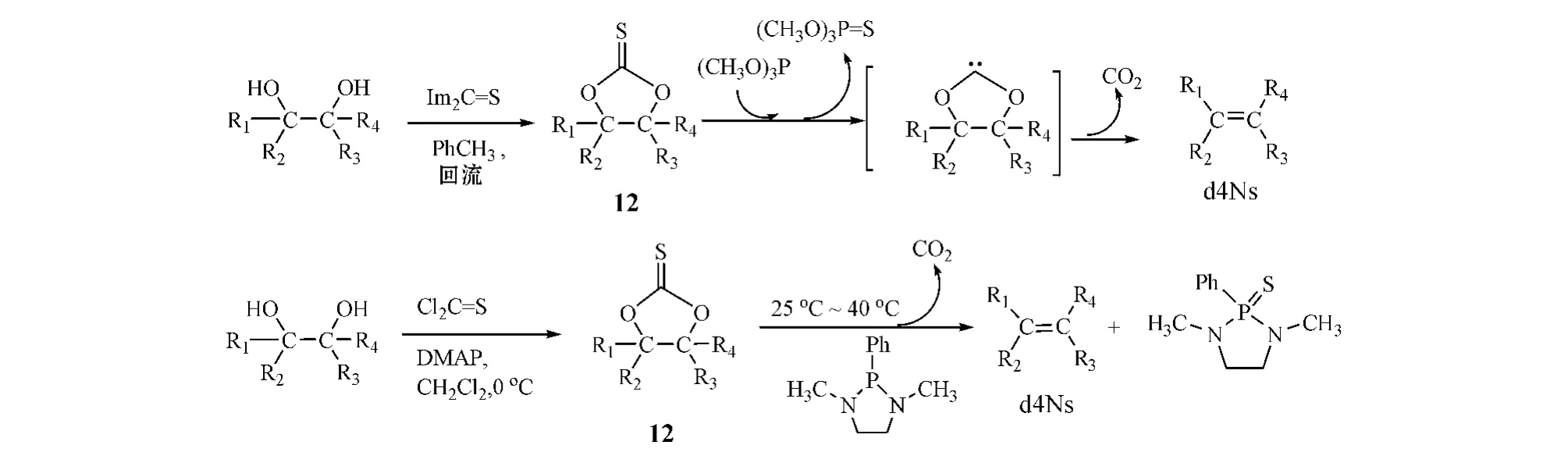
图14 用亚磷酸三甲酯或1,3-二甲基-2-苯基-1,3,2-二氮磷杂环戊烷合成d4Ns的机制Figure 14 Synthesismechanism of d4Ns using(CH3 O)3 P or 1,3-dimethyl-2-phenyl-1,3,2-diazaphospholidine
2.1.4 利用Garegg-Samuelsson反应的合成方法
Garegg-Samuelsson反应是20世纪70年代末由Garegg等创立并命名(Synthesis,1979年),即用咪唑、碘单质和三苯基膦在较温和的条件下与邻二仲羟基反应,一步即获得脱氧烯烃。于是,有人将该反应用于糖类脱氧烯烃化及合成d4Ns和ddNs(见图15)[26]。
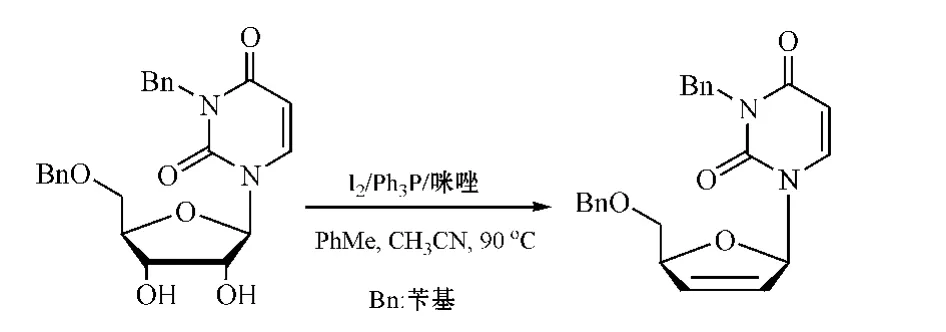
图15 利用Garegg-Samuelsson反应合成d4NsFigure 15 Synthesis of d4Ns via Garegg-Samuelsson procedure
值得注意的是,该反应较少用于嘌呤类d4Ns的合成;且用该反应用于处理嘧啶核苷时,嘧啶碱基3位NH常需用苄基保护起来,否则会发生复杂的副反应,这可从该反应所用试剂与邻二羟基化合物间的作用机制中略见一斑(见图16)[26]。
从图16可见,Garegg-Samuelsson反应实质上是利用Ph3P易被氧化和I2易被还原的性质而致糖类邻二羟基脱氧,其中咪唑起活化Ph3P的作用。

图16 Garegg-Samuelsson反应机制Figure 16 Mechanism of Garegg-Samuelsson procedure
2.1.5 利用2',3'-二-O-甲磺酰基核苷或环2',3'-硫酸酯核苷为原料的合成方法
2.1.5.1 萘基钠还原消除法 2',3'-二-O-甲磺酰基核苷或环2',3'-硫酸酯核苷是易于制备的中间体,并且甲磺酰基与环硫酸酯基都是较好的离去基团。Robins等[27]在-50℃下,利用萘基钠(sodium naphthalenide)使嘌呤核苷衍生物中2',3'-二-O-甲磺酰基或环2',3'-硫酸酯还原消除,得到d4Ns;且用环2',3'-磷酸酯核苷做原料,也可在萘基钠作用下还原消除而制得d4Ns(见图17),最后d4Ns经加氢还原得ddNs。
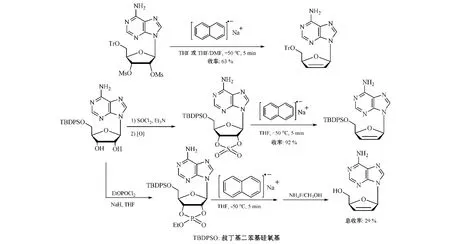
图17 经萘基钠还原消除反应合成d4NsFigure 17 Synthesis of d4Ns via reductive elimination with sodium naphthalenide
该法优点是,还原消除的收率高,且反应时间短,仅需要5min。但是,该法不适用于嘧啶核苷的脱氧反应;而且由于萘基钠稳定性差、易被氧化,溶剂需作脱水脱氧处理,反应需用惰性气体保护,反应温度低(-50℃),这些都限制了该法的推广使用。
2.1.5.2 碲/硒化钠还原消除法 Clive等[28-29]利用碲化钠(即二价碲)对嘧啶/嘌呤核苷衍生物中2',3'-二-O-甲磺酰基进行还原消除,制得d4Ns;硒与碲为同一主族,硒化钠也有相同的还原消除活性,但制得的d4Ns收率较低(见图18)。
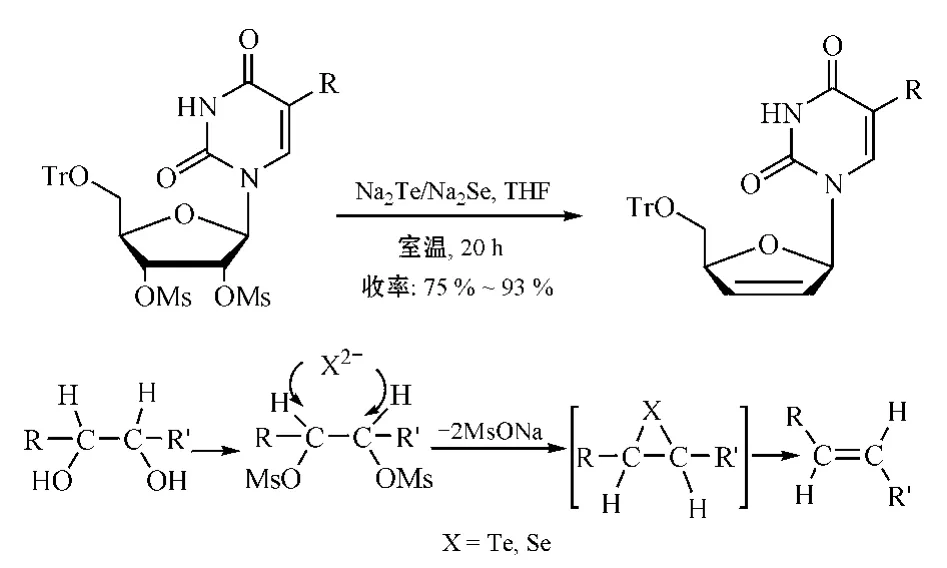
图18 经碲/硒化钠还原消除反应合成d4Ns及其机制Figure 18 Synthesis of d4Ns via reductive elimination using Na2 Te/Na2 Se and itsmechamism
碲/硒化钠因易在空气和水分中氧化,一般需现制现用,且其反应条件要求高,如需用绝对无水溶剂等,反应时间长,这些都是碲/硒化钠还原清除法的不足之处。
碲化锂对2',3'-二-O-甲磺酰基核苷也有还原消除作用(见图19),其反应条件和原理与碲化钠相同,且还原效果稍好于碲化钠[30],但其制备条件要求与价格都高于碲化钠。
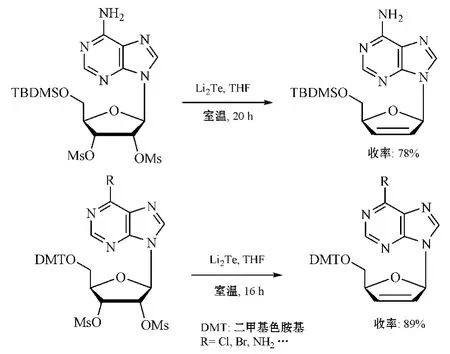
图19 经碲化锂还原消除反应合成d4NsFigure 19 Synthesis of d4Ns via reductive elimination using Li2 Te
2.1.5.3 全氟烷基联二硒酚衍生物还原消除法为克服碲/硒化钠还原消除法的不足,如较苛刻的反应条件及碲/硒化钠用后不易回收再利用等,Crich等[30]合成出一种可回收利用的还原消除催化剂——二(4-全氟己烷苯基)联二硒,即[CF3(CF2)5C6H4Se]2,其在NaBH4协同作用下,可还原消除核苷中2',3'-二-O-甲磺酰基团形成烯,所得d4Ns的收率较高(见图20)。
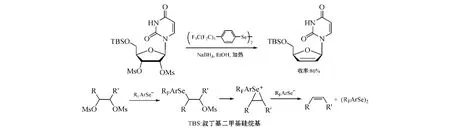
图20 经二(4-全氟己烷基苯基)联二硒还原消除反应合成d4Ns及其机制Figure 20 Synthesis of d4Ns via reductive elimination using bis(4-perfluorohexylphenyl)diselenide and itsmechanism
该法优点突出:反应条件温和,还原消除产物收率高,催化剂可回收利用。所以其有较好的应用前景。
2.2 路径1合成策略
2.2.1 利用高压加氢催化氢解-还原反应的合成方法
Antonov等[31]在含水异丙醇中,以Pd-C作催化剂,高压加氢,在较高温度下氢解-还原2',3'-二-O-甲磺酰基嘌呤核苷衍生物,获得ddNs,其收率:ddA为59%,ddI为48%(见图21)。该研究团队还证实,此氢解-还原反应不适用于2',3'-二-O-甲磺酰基嘧啶核苷衍生物;此氢解-还原反应中经历了d4Ns中间体阶段,但因反应剧烈,中间体迅速转化为最终产物ddNs。
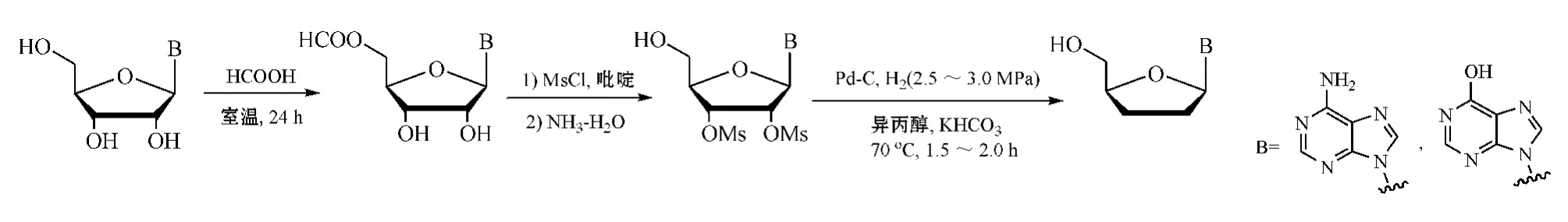
图21 经高压加氢催化氢解-还原反应合成ddNsFigure 21 Synthesis of ddNs via hydrogenolysis-reduction catalyzed by high-pressure hydrogenation
笔者所在课题组也对此法进行了验证实验,结果发现,反应中所需加氢压力要高于图21所示,否则不能发生氢解反应,因此需更好的耐压设备;其次,反应杂质多,纯化不便。这些都是该法的主要缺点,尚需进一步研究解决。
2.2.2 利用N-甲基咔唑的光感应电子转移效应引发脱氧反应的合成方法
已知N-甲基咔唑(MCZ)是一种感光剂(Saito等,JAm Chem Soc,1986年),其在高电压光源(hν,如400W高压汞灯光源等)光照和高氯酸盐作用下可产生光感应电子转移(PET)效应。于是,Wang等[32]利用该效应将MCZ与2',3',5'-三-O-苯甲酰基核苷作用,致核苷发生2',3'-脱氧反应,直接制得较高收率的ddNs(见图22)。
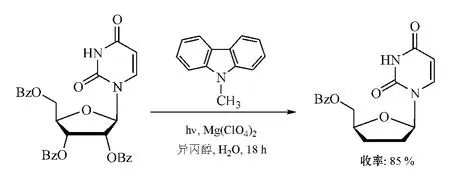
图22 利用N-甲基咔唑的光感应电子转移效应合成ddNsFigure 22 Synthesis of ddNs via the photoinduced electron transfer effect of MCZ
此法适用于各类嘧啶/嘌呤核苷作原料制备ddNs,反应前仅需对核苷的2',3'-羟基进行芳甲酰化处理,反应常以异丙醇-水为溶剂,具有用于规模化生产ddNs的潜在开发价值。
4 结语
ddNs是一类具特殊结构的活性显著的抗病毒小分子,毒性小,临床上除了用于内服治疗病毒性疾病以外,有些还可外用,且抗病毒疗效确切,其在抗病毒新药研发中占有越来越重要的地位。多年来,有关ddNs合成的方法学研究与报道较多,本文归纳了其中有代表性的、较为新颖的ddNs合成方法、途径或新试剂,以探讨ddNs合成的基本策略与规律,为合理选择ddNs合成方法提供参考和帮助。
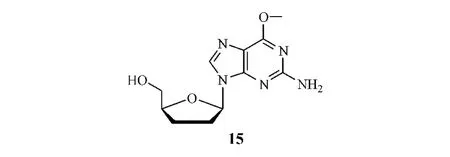
以2'-脱氧核苷或核苷为原料合成ddNs其实质就是核苷糖环上的脱氧反应,为了避免C-N糖苷键断裂,各种选择性脱氧反应都应在较温和的反应条件下进行,这是很重要的反应原则。遵循这一原则,笔者所在课题组曾应用本文所述的一种合成方法,自主研发了抗HBV新药美他卡韦(metacavir,一种2',3'-双脱氧鸟嘌呤核苷衍生物,15)肠溶胶囊,其作为1.1类新药已获我国 SFDA批准进入临床研究。
现有的ddNs合成方法与技术虽有一定开发价值,但仍存在各种缺陷与使用的局限性,除了本文中提及的以外,尚有如大规模制备时产物收率下降、制备成本较高、对原料的纯度和合成条件要求苛刻等问题,这些都有待进一步研究解决。随着相关研究的不断深入,相信今后会开发出构思更为精巧、收率高、污染少、成本合理的ddNs合成方法。
[1] Nair V.Antiviral isonucleosides:discovery,chemistry and chemical biology[M].//Chu C K.Recent Advances in Nucleosides:Chemistry and Chemotherapy.New York: Elservier Science B V,2002:149-166.
[2] Chu C K,Ma L,Olgen S,et al.Synthesis and antiviral activity of oxaselenolane nucleosides[J].J Med Chem,2000,43(21):3906-3912.
[3] Schott H,Ludwig P S,Immelmann A,et al.Synthesis and in vitro anti-HIV activities of amphiphilic heterodinucleoside phosphate derivatives containing the 2',3'-dideoxynucleosides ddC,AZT and ddI[J].Eur J Med Chem.1999,34(4):343-352.
[4] Pierra C,Imbach J L,De Clercq E,et al.Synthesis and antiviral evaluation of some beta-L-2',3'-dideoxy-5-chloro pyrimidine nucleosides and pronucleotides[J].Antiviral Res,2000,45(3):169-183.
[5] Qiu X L,Xu X H,Qing F L.Recent advances in the synthesis of fluorinated nucleosides[J].Tetrahedron,2010,66(4):789-843.
[6] 姚其正.核苷化学合成[M].北京:化学工业出版社,2005.
[7] 徐颂,姚其正.抗艾滋病药物司他呋定合成方法研究[J].药学进展,2005,29(5):203-212.
[8] Kumar V,Malhotra SV.Synthesis of nucleoside-based antiviral drugs in ionic liquids[J].Bioorg Med Chem Lett,2008,18(20):56405642.
[9] Bertolini G,Castoldi P,Deleo M,et al.Process for preparing 2',3'-didehydro-2',3'-dideoxynucleosides and 2',3'-dideoxynucleosides via reductive elimination reaction: US,10567696[P].2004-07-29.
[10]Li Z C,Chen SH,Jiang N,et al.Synthesis of triazole nucleoside derivatives[J].Nucleos Nucleot Nucleic Acids,2003,22(4):419-435.
[11]Liu L F,Li Y F,Liotta D,et al.Directed evolution of an orthogonal nucleoside analog kinase via fluorescence-activated cell sorting[J].Nucleic Acids Res,2009,37(13): 4472-4481.
[12]姜迅知,鞠玻.司他夫定的合成[J].中国医药工业杂志,2007,38(6):403-404.
[13]刘子宁,吕刚,姚其正.尿苷类酰卤化过程中相关中间体的实验研究[J].药学进展,2005,29(7):327-331.
[14]Valade A,Urban D,Beau JM.Target-assisted selection of galactosyltransferase binders from dynamic combinatorial libraries.An unexpected solution with restricted amounts of the enzyme[J].ChemBioChem,2006,7(7): 1023-1027.
[15]Katayama S,Takamatsu S,Naito M,et al.A synthesis of 3'-α-fluoro-2',3'-dideoxyadenosine via a bromine rearrangement during fluorination with MOST reagent[J].J Fluorine Chem,2006,127(4/5):524-528.
[16]Braga A L,Severo Filho W A,Schwab R S,et al.Synthesis of selenium-and tellurium-containing nucleosides derived from uridine[J].Tetrahedron Lett,2009,50(25): 3005-3007.
[17]Soares M C,de Souza M C,Ferreira V F.Strategies for the synthesis of deoxynucleosides[J].Quim Nova,2001,24(2):206-219.
[18]Chen B C,Quinlan S L,Reid JG,et al.A new thymine free synthesis of the anti-AIDS drug d4T via regio/stereo controlled-elimination of bromoacetates[J].Tetrahedron Lett,1998,39(8):729-732.
[19]Costenaro E R,Fontoura Luis A M,Oliveira D F,et al.Stereoselective synthesis of azanucleosides aza-Stavudine (aza-D4T),aza-2',3'-didehydro-3'-deoxyuridine(aza-D4U),and its hydrogenated analogues from an endocyclic enecarbamate[J].Tetrahedron Lett,2001,42(9): 1599-1602.
[20]Chen B C,Quinlan SL,Stark D R,etal.5'-Benzoyl-2'-αbromo-3'-O-methanesulfonylthymidine:a superior nucleoside for the synthesis of the anti-AIDS drug d4T(stavudine)[J].Tetrahedron Lett,1995,36(44):7957-7960.
[21]靳立人,蒋洪平,侯鹏翼.司他夫定的合成[J].厦门大学学报,2002,41(2):207-210.
[22]Hiroshi S,Yusuke A,Yutaka H,et al.Synthesis of 2',3'-dideoxypurinenucleosides via the palladium catalyzed reduction of 9-(2,5-di-O-acetyl-3-bromo-3-deoxy-D-xylofuranosyl)purine derivatives[J].Nucleos Nucleot,1996,15(1/3):31-45.
[23]Chun R E,Vadakkan J J,Nair V.Ring-expanded analogues of natural Oxetanocin:(+)-and(-)-hydroxymethyl isodideoxyadennosine[J].Nucleos Nucleot Nucleic Acids,2005,24(5):725-728.
[24] Torii T,Izawa K,Cho D H,et al.Synthesis of 2',3'-dideoxyinosine via radical deoxygenation[J].Nucleos Nucleot Nucleic Acids,2007,26(8/9):985-988.
[25]Nguyen C,Kasinathan G,Gilbert IH,et al.Deoxyuridine triphosphate nucleotidohydrolase as a potential antiparasitic drug target[J].JMed Chem,2005,48(19):5942-5954;
[26]Kostina V G,Alexeeva IV,Palchikovskaya,L I.Synthesis of novel 2',3'-dideoxy-6-azacytidine derivativespotential retroviral reproduction inhibitors[J].BiopolymCell,2001,17(6):560-564.
[27] Robins M J,Lewandowska E,Wnuk S F.Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides from ribonucleoside cyclic 2',3'-(sulfates or phosphates)or 2',3'-dime-sylates via reductive elimination with sodium naphthalenide[J].JOrg Chem,1998,63(21):7375-7381.
[28]Clive D L J,Wickens P L,Sgarbi PW M.Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides by reaction of 5'-protected nucleoside 2',3'-dimesylates with telluride dianion:a general route from cis vicinal diols to olefins[J].JOrg Chem,1996,61(21):7426-7437.
[29]Clive D L J,Sgarbi PW M,Wickens P L.Synthesis of 2',3'-Didehydro-2',3'-dideoxynucleosides by reaction of 5'-O-protected nucleoside 2',3'-dimesylates with lithium areneselenolates[J].JOrg Chem,1997,62(11): 3751-3753.
[30]Crich D,Neelamkavil S,Sartillo-Piscil F.Efficient conversion of vicinal diols to alkenes by treatment of the corresponding dimesylates with a catalytic,minimally fluorous,recoverable diaryl diselenide and sodium borohydride[J].Org Lett,2000,2(25):4029-4031.
[31]Antonov K V,Konstantinova ID,Miroshnikov A I.New approach to the synthesis of 2',3'-dideoxyadenosine and 2',3'-dideoxyinosine[J].Nucleos Nucleot,1998,17(1/ 3):153-159.
[32]Wang ZW,Prudhomme D R,Rizzo C J,et al.Stereocontrolled syntheses of deoxyribonucleosides via photoinduced electron-transfer deoxygenation of benzoyl-protected riboand arabinonucleosides[J].JOrg Chem,2000,65(19): 5969-5985.
