吲哚并喹啉类似物的合成及抗肿瘤活性评价*
2011-01-08孟俊秀万升标任素梅
孟俊秀,万升标,任素梅,江 涛
(中国海洋大学医药学院,教育部海洋药物重点实验室,山东青岛266003)
吲哚并喹啉类似物的合成及抗肿瘤活性评价*
孟俊秀,万升标,任素梅,江 涛**
(中国海洋大学医药学院,教育部海洋药物重点实验室,山东青岛266003)
建立了1种合成吲哚并喹啉羧酸化合物简便有效的方法。以邻氨基苯甲酸为起始原料,经酰化、缩合、环和、氯代、水解、还原多步反应得到6个吲哚并喹啉类似物。所合成化合物结构均经核磁共振及质谱技术验证,并利用MTT法测定了这6个吲哚并喹啉类似物对人乳腺癌细胞株MDA-231的体外扩增抑制活性。
吲哚并喹啉类似物;抗肿瘤活性;合成
天然化合物吲哚并喹啉(quindoline)与白叶藤碱(crytolepine)是从非洲灌木Cryptolepis sanguinolenta的根中提取分离出来的2种吲哚并喹啉类生物碱[1]。现代生物学实验发现白叶藤碱具有抗炎、抗菌及抗肿瘤活性[2]。白叶藤碱之所以具有抗肿瘤活性是由于其不仅具有DNA插入活性[3],而且具有较好的拓扑异构酶Ⅱ[4]和端粒酶抑制活性[5]。其端粒酶抑制活性主要是通过稳定端粒酶中的G-四螺旋结构而表现出来的,对其进行结构改造,得到了一系列活性较好的端粒酶抑制剂[5-7]。目前对吲哚并喹啉的结构改造主要是在A环11-位引入各种烷基胺侧链[8]、芳香基团[9]、羧酸官能团[10],C环10-位引入亲脂侧链[9],A环和B环引入卤素、甲基、硝基等官能团[11]。目前,仍未发现通过向D环9-位引入羧基对其进行结构改造及生物活性评价的报道。因此本文欲设计合成一系列D环9-位羧基取代的吲哚并喹啉的类似物,并对其进行体外抗肿瘤活性的测试评价。
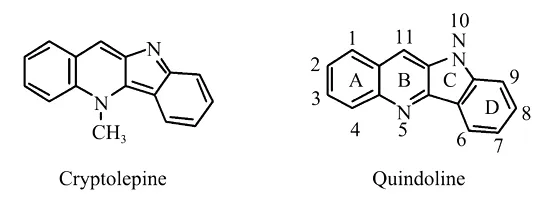
图1 吲哚并喹啉及白叶藤碱的结构[1]Fig.1 Structures of quindoline and crytolepine
1 实验部分
1.1 仪器与试剂
JEOL JNM-EPC 600 NMR核磁共振仪(内标TMS);Q-TOF Ultima TM Global质谱仪;所用试剂均为市售化学纯或分析纯。MDA-231肿瘤细胞株扩增抑制活性采用标准MTT法测得。
1.2 合成方法
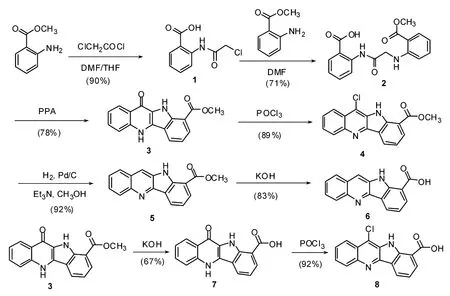
1.2.1 2-[(2-氯乙酰)氨基]-苯甲酸(1) 向盛有200 mL DMF的三颈瓶中加入邻氨基苯甲酸(16.46 g,120.00 mmol),磁力搅拌使之溶解。在冰浴下,向上述溶液滴加氯乙酰氯(14.91 g,132.03 mmol)的四氢呋喃(50 m L)溶液,控温低于25℃。滴加完毕后撤去冰水浴,搅拌反应24 h。将反应液滴加到1 000 m L水中,并使用机械搅拌,此时有大量白色固体析出。抽滤,得白色固体,干燥后经硅胶柱层析(乙酸乙酯∶石油醚=1∶1)纯化,得化合物1(23.13 g,90%)。1HNMR(DMSO-d6,600 MHz):δ13.80(1H,bs,COO H),11.84(1H,bs,N H),8.54(1H,d,J=7.8 Hz,Ar H),8.03(1H,dd,J=7.8 Hz,J=1.9 Hz,Ar H),7.66-7.63(1H,m,Ar H),7.24-7.21(1H,m,Ar H),4.47(1H,s,CH2)ppm.13CNMR(DMSO-d6,151 MHz):δ169.9,165.8,140.5,134.8,131.8,124.0,120.4,117.4,44.0;TOF MS(ES-):m/z(relative intensity),212.0(100%)[M-H]-;HRMS(ES-):m/z[M-H]-calcd for C9H7Cl NO3:212.0114;found 212.0107。
1.2.2 2-[2-[2-(甲氧羰基)苯基胺]乙酰氨基]苯甲酸(2) 向100 m L三口圆底烧瓶中加入2-[(2-氯乙酰)氨基]-苯甲酸(1)(6.73 g,31.50 mmol),加入25 m L DMF搅拌使其溶解。然后加入邻氨基苯甲酸甲酯(29.20 g,19.32 mmol),氮气保护下于92℃油浴反应60 h。将反应液倒入150 m L水中,充分搅拌,弃去上清液后,加入150 m L乙酸乙酯使生成的粘稠物溶解。分批加入100 m L质量分数为6%的碳酸氢钠水溶液,充分搅拌使产物形成钠盐沉淀出来,抽滤,滤饼用30 m L乙酸乙酯洗3次,将滤饼分散到100 m L四氢呋喃中,用2 mol/L盐酸中和到p H=5,沉淀消失,分去水层,有机相以饱和食盐水洗涤2次后,以无水硫酸镁干燥,抽滤后,将滤液减压浓缩得粘稠物。加入适量四氢呋喃回流使粘稠物溶解,然后滴加石油醚,有白色固体沉出,抽滤干燥得化合物2(7.30 g,71%)。1HNMR(DMSO-d6):δ13.53(1 H,s,N H),11.82(1 H,s,N H),8.68(1H,dd,J=8.7,1.4 Hz,Ar H),8.25(1H,t,J=5.9 Hz,Ar H),7.94(1H,dd,J=7.8,1.4 Hz,Ar H),7.86(1 H,dd,J=8.3,1.9 Hz,Ar H),7.59(1 H,td,J=8.7,1.8 Hz,Ar H),7.13(1 H,td,J=7.3,1.4 Hz,Ar H),6.68(1H,td,J=8.3 Hz,1.0 Hz,Ar H),6.61(1H,d,J=8.2 Hz,Ar H),4.12(2H,d,J=5.9 Hz,CH2),3.84(s,3H,OCH3);TOF MS(ES):m/z(relative intensity),327.0(100%)[M H]-;HRMS:m/z[M H]-calcd.for C17H15N2O5:327.098 1,found 327.097 9。
1.2.3 5,11-二氢-11-氧代-10 H-吲哚[3,2-b]喹啉-9-羧酸甲酯(3) 在50 m L的两口瓶中加入化合物2(2.00 g,6.10 mmol),多聚磷酸(55 g,16.27 mmol),使用玻璃棒搅拌使之混合均匀,在100℃油浴上反应2 h,反应完全。将反应液趁热倒入到200 m L冰水中,充分搅拌,离心得黄色泥状固体。水洗之后再次离心,重复此操作3次。黄色泥状固体溶于50 m L四氢呋喃中,20 m L饱和氯化钠水溶液洗2次,无水硫酸镁干燥,将四氢呋喃浓缩,大量黄色固体生成,抽滤,真空干燥,得黄色固体3(1.40g,78%)。1HNMR(DMSO-d6):δ12.65(1H,s,N H),10.57(1H,s,N H),8.53(1H,d,J=7.8 Hz,Ar H),8.37(1H,d,J=7.8 Hz,Ar H),8.15(1H,dd,J=7.8,0.96 Hz,Ar H),7.75-7.71(2H,m,Ar H),7.40(1H,t,J=7.7 Hz,Ar H),7.34(1H,t,J=7.8 Hz,Ar H),4.01(3H,s,OCH3);13CNMR(DMSO-d6):δ167.22,166.31,139.22,136.45,131.17,129.88,129.04,126.79,125.21,123.22,122.87,120.98,119.02,118.20,117.93,113.51,52.30;TOF MS(ES+):m/z(relative intensity),293.1(100%)[M+H]+;HRMS:m/z[M+H]+calcd.for C17H13N2O3:293.092 6,found 293.092 8。
1.2.4 11-氯-10 H-吲哚[3,2-b]喹啉-9-羧酸甲酯(4)
在250 m L单口瓶中加入化合物3(1.02 g,3.49 mmol),三氯氧磷60 m L,回流反应18 h。常压下蒸除大部分三氯氧磷,剩余反应液与冰水充分搅拌,离心得到黄色沉淀。水洗之后再次离心,重复此操作3次。真空干燥得到化合物4(0.97 g,89%)。1HNMR(DMSO-d6,600 MHz):δ11.65(s,1H,N H);8.66(1H,d,J=7.8 Hz,Ar H),8.35(1H,dd,J=8.2,0.9 Hz,Ar H),8.31(1 H,dd,J=7.8,0.90 Hz,Ar H),8.25(1H,dd,J=7.8,0.9 Hz,Ar H),7.83(1H,m,Ar H),7.80(1H,m,Ar H),7.50(1H,t,J=7.3 Hz,Ar H),4.04(3H,s,OCH3);13CNMR(DMSO-d6,151 MHz):δ166.1,145.1,144.5,142.7,131.6,129.9,129.4,127.7,127.0,123.9,123.3,122.5,120.5,119.4,113.2,52.42.TOF MS(ES+):m/z(relative intensity),311.1(100%)[M+H]+;HRMS(ES+):m/z[M+H]+calcd for C17H12Cl N2O2:311.058 7;found 311.057 5。
1.2.5 10 H-吲哚[3,2-b]喹啉-9-羧酸甲酯(5) 于50 mL单口瓶中加入化合物4(0.85 g,2.74 mmol),加入50 m L四氢呋喃/甲醇(1∶1)混合溶剂,10%钯碳(0.40 g),三乙胺(0.5 m L),通入氢气,搅拌反应24 h,滤去钯碳,减压浓缩得黄色固体,四氢呋喃/甲醇中重结晶,得化合物5(0.70g,92%)。1HNMR(DMSO-d6,600 MHz)δ:11.98(1H,s,NH);8.91-8.77(2H,m,Ar H);8.38-8.31(3H,m,Ar H),7.87(1 H,d,J=5.94 Hz,Ar H),7.73(t,J=5.94 Hz,1H,Ar H),7.49(1 H,t,J=7.3 Hz,Ar H),4.04(3 H,s,OCH3);13CNMR(DMSO-d6,151 MHz):δ165.8,142.9,133.17,132.4,128.3,127.9,126.6,126.0,119.6,112.7,52.3;TOF MS(ES+):m/z(relative intensity),277.1(100%)[M+H]+;HRMS(ES+):m/z[M+H]+calcd for C17H13N2O2:277.097 7;found 277.097 0。
1.2.6 10 H-吲哚[3,2-b]喹啉-9-羧酸(6) 向三口烧瓶中分别加入KOH(3.30 g,58.81 mmol)、H2O(50 m L)以及化合物5(0.70 g,2.53 mmol)。混合物搅于40℃下,拌反应8 h直至形成澄清溶液。使用2 mol/L HCl将p H值调整为6,大量黄色沉淀形成,抽滤,水洗,干燥得黄色固体6(0.55 g,83%)。1HNMR(DMSO-d6,600 MHz):δ11.72(1H,s,NH),8.75(1H,d,J=6.4 Hz,Ar H),8.62(1H,s,Ar H),8.30(1H,d,J=8.3 Hz,Ar H),8.26(1H,d,J=7.8 Hz,Ar H),8.2(1H,d,J=8.3 Hz,Ar H),7.79(1H,t,J=6.9 Hz,Ar H),7.66(1H,t,J=7.3 Hz,Ar H),7.44(1H,t,J=7.8 Hz,Ar H);13CNMR(DMSO-d6,151 MHz):δ167.4,143.2,133.0,132.0,128.0,127.4,126.8,126.7,125.6,119.2,113.5;TOF MS(ES+):m/z(relative intensity),263.1(100%)[M+H]+;HRMS(ES+):m/z[M+H]+calcd for C16H11N2O2:263.082 1;found 263.080 9。
1.2.7 5,11-二氢-11-氧代-10 H-吲哚[3,2-b]喹啉-9-羧酸(7) 制备方法同化合物6。产率:67%。1HNMR(DMSO-d6,600 MHz):δ13.64(1 H,bs,COO H),12.68(1H,s,N H),10.22(1H,s,CON H),8.51(1 H,d,J=7.8 Hz,Ar H),8.37(1H,d,J=7.8 Hz,Ar H),8.13(1H,dd,J=7.3,1.0 Hz,Ar H),7.71-7.76(2 H,m,Ar H),7.38(1H,t,J=7.8 Hz,Ar H),7.35-7.33(1H,m,Ar H);13CNMR(DMSO-d6,151 MHz):δ168.5,167.7,139.8,137.8,131.7,130.4,126.9,125.7,123.6,121.6,119.6,118.6,118.4;TOF MS(ES+):m/z(relative intensity),279.1(100%)[M+H]+;HRMS(ES+):m/z[M+H]+calcd for C16H11N2O3:279.077 0;found 279.077 2。
1.2.8 11-氯-10 H-吲哚[3,2-b]喹啉-9-羧酸(8) 合成方法同化合物4。产率:92%。1HNMR(DMSO-d6,600 MHz):δ10.43(1H,s,N H),8.59(1H,d,J=7.3,1.4 Hz,Ar H),8.30-8.27(2H,m,Ar H),8.22(1H,d,J=1.3 Hz,Ar H),7.82-7.79(1H,m,Ar H),7.78-/Z M7.75(1H,m,Ar H),7.46(1H,t,J=7.3 Hz,Ar H);13CNMR(DMSO-d6,151 MHz):δ167.7,145.1,144.4,143.2,131.7,129.6,129.4,127.5,126.9,126.4,123.8,122.9,122.4,120.4,119.10 TOF MS(ES+):m/z(relative intensity),297.0(100%)[M+H]+;HRMS(ES+):m/z[M+H]+calcd for C16H10Cl N2O2:297.043 1;found 297.044 1。
2 结果与讨论
2.1 合成方法
实验室在合成四环体系羧酸化合物的研究中发现,环合反应的底物如果不使用甲基保护其中的1个羧酸,难以分离纯化。而且,此类底物若为双羧酸体系在环合反应时容易形成大环三聚物[12]。因此,本文通过改进已有吲哚并喹啉四环体系的合成方法[13],以甲酯化合物2为环合反应的底物,建立了1种吲哚并喹啉羧酸(6)的合成方法,并通过该方法合成了另外5个吲哚并喹啉的类似物,即化合物3,4,5,7,8。各步产率较高,易于制备。首先,以邻氨基苯甲酸为起始原料与氯乙酰氯反应得到2-[(2-氯乙酰)氨基]-苯甲酸(1);2-[(2-氯乙酰)氨基]-苯甲酸(1)与邻氨基苯甲酸甲酯发生缩合反应得到化合物2;化合物2在多聚磷酸(PPA)中环合得到具有四环体系的羧酸甲酯3;化合物3与三氯氧磷发生氯代反应得到化合物4;化合物4由钯碳催化氢化可以得到化合物5;化合物5在氢氧化钾水溶液中水解可制得吲哚并喹啉羧酸(6)。吲哚并喹啉羧酸(6)的制备共经历6歩反应,总产率为34%。具有四环体系的羧酸化合物7和8可以通过类似方法以高产率制得,合成路线如下:
2.2 抗肿瘤活性评价
本文使用人乳腺癌细胞株MDA-231,在浓度为5μmol/L下对所合成的6个吲哚并喹啉类似物利用MTT法进行了抗肿瘤生物活性体外筛选,结果见表1。活性测试结果表明合成的吲哚并喹啉类似物中,羧酸甲酯化合物3,4,5在浓度为5μmol/L时对乳腺癌细胞株的增值具有一定的抑制活性,而其对应的羧酸化合物7,8,6的活性则相对较差。这可能是由于羧酸甲酯化合物相对于羧酸化合物,其脂溶性较好易于被细胞吸收所致。另外,根据文献报道,在吲哚并喹啉A环11-位[8]、C环10-位[9]引入的烷基胺侧链上的氮原子在细胞内可以形成正电荷中心,有利于稳定端粒酶上的G-四螺旋结构,从而提高该类化合物的端粒酶抑制活性。本文所合成的吲哚并喹啉类似物的B环11-位、C环10-位及D环9-位易于进行结构修饰,特别是可以通过羧酸官能团与各种脂肪胺形成酰胺键,从而向D环引入各种烷基胺侧链。因此,实验室下一步的工作即向吲哚并喹啉A环、C环及D环引入各种烷基胺侧链,并对其进行构效关系分析。

表1 吲哚并喹啉类化合物对人乳腺癌细胞株MDA-231的扩增抑制活性Table 1 Proliferation inhibition of indoquinoline compounds(5μmol/L)against breast cancer cell line MDA-231
[1] Dwuma-Badu D,Ayin J S K,Fiagbe N I,et al.Constituents of west african medicinal plants XX:quindoline from Cryptolepis sanguinolenta.[J].J Pharm Sci,1978,67:433-434.
[2] Dassonneville L,Lansiaux A,Wattelet A,et al.Cytotoxicity and cell cycle effects of the plant alkaloids cryptolepine and neocryptolepine:relation to drug induced apoptosis.[J].Eur J Pharmacol,2000,409:9-18.
[3] Lisgarten J N,Coll M,Portugal J,et al.The antimalarial and cytotoxic drug cryptolepine intercalates into DNA at cytosine cytosine sites.[J].Nature Structural Biology,2002,9:57-60.
[4] Bonjean K,De Pauw Gillet M C,Defresne M P,et al.The DNA intercalating alkaloid cryptolepine interferes with topoisomerase II and inhibits primarily DNA synthesis in B16 melanoma cells[J].Biochemistry,1998,37:5136-5146.
[5] Guyen B,Schultes C M,Hazel P,et al.Synthesis and evaluation of analogues of 10 H indolo[3,2 b]quinoline as G quadruplex stabilising ligands and potential inhibitors of the enzyme telomerase[J].Org Biomol Chem,2004,2:981-988.
[6] Zhou J L,Lu Y J,Ou T M,et al.Synthesis and evaluation of quindoline derivatives as G quadruplex inducing and stabilizing ligands and potential inhibitors of telomerase[J].J Med Chem,2005,48:7315-7321.
[7] Lu Y J,Ou T M,Tan J T,et al.5-N Methylated quindoline derivatives as telomeric G quadruplex stabilizing ligands:effects of 5-N positive charge on quadruplex binding affinity and cell proliferation[J].J Med Chem,2008,51:6381-6392.
[8] Caprio V,Guyen B,Opoku Boahen Y,et al.A novel inhibitor of human telomerase derived from 10 H indolo[3,2 b]quinoline[J].Bio Med Chem Lett,2000,10:2063-2066.
[9] Yamato M,Takeuchi Y,Chang M R,et al.Synthesis and antitumor activity of fused quinoline derivatives[J].Chem Pharm Bull,1990,38:3048-3052.
[10] Yang S W,Abdel-Kader M,Malone S,et al.Synthesis and biological evaluation of analogues of cryptolepine,an alkaloid isolated from the suriname rainforest[J].J Nat Prod,1999,62:976-983.
[11] Onyeibor O,Croft S L,Dodson H I,et al.Synthesis of some cryptolepine analogues,assessment of their antimalarial and cytotoxic activities,and consideration of their antimalarial mode of action[J].J Med Chem,2005,48:2701-2709.
[12] Wan S B,Liu Z L,Chen D,et al.Polyphosphorous acid catalyzed cyclization in the synthesis of cryptolepine derivatives[J].Chinese Chem Lett,2007,18:1179-1181.
[13] Sunder S,Peet N P.Syntheis of benzofuro[3,2-b]quinolin-6(11H)one and derivatives[J].J Heterocyclic Chem,1978,15:1379-1382.
Synthesis and Antitumor Activity Evaluation of Quindoline Analogues
MENG Jun-Xiu,WAN Sheng-Biao,JIANG Tao
(The Key Laboratory of Marine Drugs,Ministry of Education,College of Medicine and Pharmacy,Ocean University of China,Qingdao 266003,China)
Abatract:A facile method for a series of indoquinoline carboxylic acids was established.Six analogues of quindoline were synthesized using anthranllic acid as starting material via acylation,condensation,cyclization,chlorination,hydrolysis and hydrogenation.All synthesized compounds were confirmed by NMR and MS techniques.Proliferation inhibitory activity against the human breast cancer cell line(MDA 231)in vitro was investigated by using standard method of MTT.
analogues of quindoline;antitumor activity;synthesis
R914.5
A
1672-5174(2011)11-091-04
科技部国际科技合作项目-CMST(2008DFA31040)资助
2011-01-10;
2011-09-29
孟俊秀(1981-),男,博士生。E-mail:jm3y07@googlemail.com
**通讯作者:E-mail:jiangtao@ouc.edu.cn
责任编辑 徐 环
