氢原子在Be(0001)表面吸附的密度泛函理论研究
2010-12-05陶向明王芒芒蔡建秋谭明秋
宁 华 陶向明 王芒芒 蔡建秋 谭明秋
(浙江大学物理系,杭州 310027)
氢原子在Be(0001)表面吸附的密度泛函理论研究
宁 华 陶向明 王芒芒 蔡建秋 谭明秋*
(浙江大学物理系,杭州 310027)
采用第一性原理的密度泛函理论研究单个氢原子和多个氢原子在Be(0001)表面吸附性质.给出了氢吸附Be(0001)薄膜表面的原子结构、吸附能、饱和度、功函数、偶极修正等特性参数.同时也讨论了相关吸附性质与氢原子覆盖度(0.06-1.33 ML)的关系.计算结果表明:氢原子的吸附位置与覆盖度之间有强烈的依赖关系,覆盖度低于0.67 ML时,氢原子能量上易于占据fcc或hcp的中空位置;覆盖度为0.78 ML时,中空位与桥位为氢原子的最佳吸附位;覆盖度在0.89到1.00 ML时,桥位是氢原子吸附能量最有利的位置;以上覆盖度中Be(0001)表面最外层铍原子的结构均没有发生明显变化.当覆盖度为1.11-1.33 ML,高覆盖度下Be(0001)表面的最外层铍原子部分发生膨胀,近邻氢原子渗入到铍表面次层,氢原子易于占据在hcp和桥位.吸附结构中的氢原子比氢分子中的原子稳定.当覆盖度大1.33 ML时,计算结果没有发现相对于氢分子更稳定的吸氢结构.同时从分析偶极修正和氢原子吸附垂直高度随覆盖度的变化关系判断氢覆盖度为1.33 ML时,在Be(0001)表面吸附达到饱和.
密度泛函理论;Be(0001)表面;氢原子吸附;吸附能
由于过渡金属在催化技术领域的应用,使得氢吸附过渡金属表面的研究受到了人们的广泛关注.目前基于密度泛函理论的第一性原理方法对氢吸附多种过渡金属(钨、镍、钯和钌)表面进行了研究[1-6].在材料结构、表面薄膜电子结构性质等研究中,氢吸附简单金属也受到人们的重视.其中,氢与铍表面的相互作用在核聚变反应堆中是非常重要的课题.尽管同为简单金属,铍的电子结构与周期表中相邻的其他元素相比有显著不同.其体相具有类半导体性质,而Be(0001)表面表现出典型的自由电子金属行为.同时,铍是原子反应堆中快速中子的主要来源[7],并且在氢燃料电池储氢材料、内燃机及在能量交换器等器件上有重要的运用[8],因此研究氢吸附Be(0001)表面的课题在实验和理论上都具有重要的意义.
实验上,人们早期研究了氢气吸附多晶铍和氢气、原子氢、原子氘吸附Be(0001)表面等性质[9-10].最近,Doerner[11]和Reinelt[12]等运用国际核聚变实验堆(ITER)中不同的位置和程序升温脱附(TPD)实验模拟了氢与铍的相互作用.到目前为止,实验上报道氢吸附Be(0001)表面的工作还是很少的.理论上,Yu等[13]于较早时间采用第一性原理的HedinLunqvist[14]交换关联赝势和平面波方法研究了氢吸附Be(0001)表面的性质.他们得出桥位是氢吸附铍表面的稳定吸附位,并给出清洁Be(0001)表面和吸附表面的相关性质.随后,Marino等[15]基于Be45团簇结构,运用第一性原理的Hartree-Fock方法模拟氢吸附Be (0001)团簇结构.事实上,团簇方法由于周期性边界条件的限制不适合用来研究表面问题.接下来, Feibelman[16-17]和Stumpf[18]等考虑了周期性边界条件,采用线性缀加平面波(LAPW)和局域梯度近似(LDA)的交换关联势的方法计算得到一系列氢吸附Be(0001)表面的结果.他们研究得到,低覆盖度(单层,ML)下氢原子易于占据在六角位(hcp),高覆盖度下(2/3-1.0 ML)新奇的氢表面空缺结构为主要吸附结构,并分析了honeycomb结构的重构[16].Feibelman[17]得出原子氢吸附在Be(0001)表面以外还能占据在次表面.Stumpf[18]则主要讨论了增强的氢原子迁移率和Be(0001)表面缺陷的形成.
最近Allouche[19]采用QUANTUM ESPRESSO软件包的超软赝势(USPP)方法对氢吸附Be(0001)表面进行了理论研究.他们选取了p(3×3)周期表面作为研究对象,选择了两种初始模型:分别固定吸附原子的xH,yH,zH坐标或是固定氢原子zH(氢原子吸附垂直高度).事实上,文献中固定吸附原子高度的计算并不能合理地解释氢吸附Be(0001)表面的物理过程,尤其在研究多个氢原子的吸附问题时.如果计算中考虑释放吸附原子坐标以及偶极磁矩修正,那么相应的研究结果将会更合理和精确.因此,本文选择释放所有吸附原子的坐标,采用密度泛函理论的投影叠加波(PAW)总能计算了0.06-1.33 ML下氢吸附Be(0001)表面的性质,包括吸附结构、吸附能、功函数、偶极修正等物理量,得到一系列新的结果并讨论了相关物理量随氢原子覆盖度的变化关系.
1 计算方法
本文使用的密度泛函自洽计算采用了维也纳从头计算程序包[20](VASP).这是一个第一性原理的量子力学分子动力学程序包,采用超软赝势[21-22]或投影缀加波(PAW)[23]并以平面波为基函数进行总能和电子结构的计算.计算中交换关联能部分包含了由Perdew、Burke和Ernzerhof提出的广义梯度近似[24-25]文献中称为GGA 96或GGA-PBE.这里在计算结构时选用了七层铍原子构成层晶结构来模拟表面.其中最低四层作为衬底固定,而其余的三层铍原子与外层吸附的氢原子在结构优化计算中是可以变化的,用来模拟表面原子的弛豫.在表面结构优化的计算中,没有考虑自旋极化的影响,但是考虑了偶极修正的作用,并在Z方向周期性排列的相邻层晶之间留有厚度1 nm的真空区域,以避免层晶之间的干扰.我们对这些设置进行了必要的数值检验,结果表明这个结构模型足以保证计算的精确度,又不至于使计算量过于庞大.
2 计算结果及分析
2.1 块体Be结构和清洁的Be(0001)表面
在研究氢原子吸附性质之前,先来研究一下块体Be结构和清洁的Be(0001)表面的表面弛豫.根据优化计算,得到的大块六角密积(hcp)结构晶体铍的晶格参数 a=0.227 nm,c/a=1.57,与实验值 a= 0.229 nm,c/a=1.569[28]相比理论值a略小1.7%,而理论值的与测量值几乎一致.此外以七层Be(0001) p(1×1)周期层晶模型模拟清洁的Be(0001)表面并计算了清洁表面的弛豫.与其它相关理论和实验的结果(见表1[16,29-32])相比,文献[30]中得到的结构弛豫特性(Δd12/d0=+5.9%)与实验值最为接近.然而,文献[30]采用的多体有效势和团簇结构的计算方法由于其处理边界条件的局限性使得它不适合用来研究周期表面的问题.我们得到的Be(0001)p(1×1)周期表面的原子弛豫Δd12/d0为+3.38%,与实验测量值相比要小一些((5.8±0.4)%).表1显示计算结果与实验值基本相符.有理由认为,使用的计算方法和结果在目前的密度泛函理论的框架下都是可信的.下文将着重讨论有关Be(0001)表面吸附氢原子的计算.
2.2 氢吸附Be(0001)表面
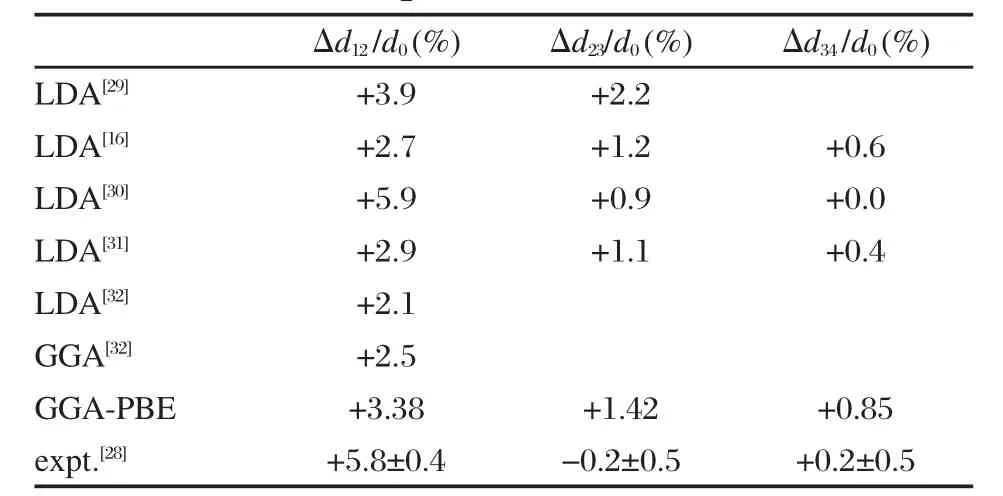
表1 清洁Be(0001)p(1×1)周期表面原子结构弛豫特性与文献值的比较Table 1 Comparison of calculated surface relaxation of Be(0001)p(1×1)with related reference and experiment data
计算氢原子吸附能时采用了如下公式:
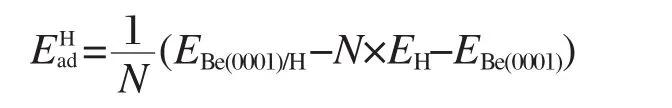
其中N为氢原子个数,EBe(0001)/H、EH和EBe(0001)分别为吸氢Be(0001)表面结构、氢原子及Be(0001)表面结构的总能.氢原子的吸附能也可以表示为

EH2是氢分子的总能.考虑了自旋极化,我们得到氢分子的结合能和键长分别约为4.53 eV和0.075 nm,与相应的实验值(4.74 eV,0.074 nm)基本符合.
2.2.1 单个氢吸附Be(0001)表面
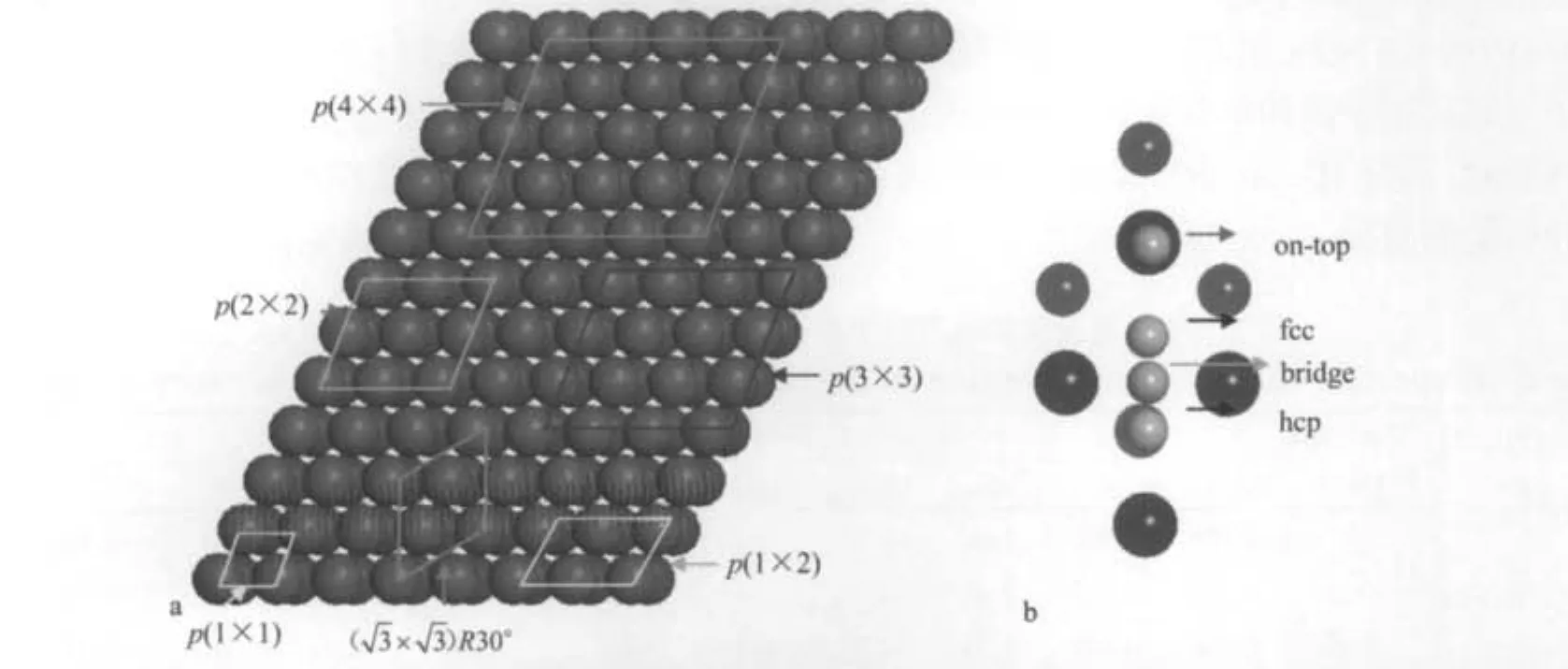
图1 六种Be(0001)周期表面结构(a)和四种氢原子吸附位置(b)中上位(top)、面心(fcc)、六角(hcp)和桥位(bridge)示意图Fig.1 Six different surface unit cells of Be(0001)surface(a)and four on-surface adsorption sites for single H atom adsorption on Be(0001)surface(b)including fcc,hcp,bridge,and on-top sitesBeryllium atoms of outmost two layers are shown by scaled big balls,hydrogen atoms are shown using small balls.
首先研究单个氢原子吸附Be(0001)表面的情况.讨论单个原子吸附金属表面可得到局域态下吸附原子与表面原子间的相互作用等性质,这些低覆盖度下的系统讨论为高覆盖度扩展态下的吸附问题提供了参考.为了确定单个氢原子吸附的最佳位置,采用了6种周期超原胞考察了四种可能的吸附位置,分别是顶位(top),六角位(hcp),桥位(bridge)和面心位(fcc).计算得到p(1×1)周期超原胞(1.0 ML)不同吸氢位置的吸附能分别为-2.15 eV(top),-2.20 eV (fcc),-2.08 eV(hcp)和-2.27 eV(bridge).结果表明桥位是原子氢在Be(0001)表面的最佳吸附位置,与文献[33]相符.需要指出的是这个桥位并不是传统意义上的桥位,它距离桥中心0.005 nm,偏向fcc的位置.同时优化得到的近桥位氢原子与最近邻铍原子的键长dH—Be(0.148 nm),氢原子距离Be(0001)表面的吸附垂直高度h(0.095 nm)以及功函数Φ(3.20 eV)均与文献[16]符合得很好.当覆盖度逐次降低为0.5、0.33、0.25、0.11和0.06 ML时,单个氢原子吸附Be(0001)表面的最佳吸附位为hcp位.其它吸附位,例如桥位和上位都不能稳定存在,而fcc位是一个局域稳定吸附位.
表2给出了不同覆盖度时单个氢原子吸附Be(0001)表面hcp和fcc位的相关性质.通过比较发现,氢原子占据hcp位相应的吸附垂直高度以及吸附能均比fcc位占优,说明hcp位确实是单个氢原子在低覆盖度下的最佳吸附位置.其中在0.33 ML情况下,氢吸附Be(0001)表面hcp位的相关性质(dH—Be、h、Φ等)均与文献[16]取得一致.表2数据表明,单个氢原子吸附Be(0001)表面的功函数会随着覆盖度的增加而逐渐减少,从5.23 eV(约0.06 ML)到3.60 eV(约0.5 ML).我们认为这是一部分电子在氢原子与基底原子间的电荷转移引发较大表面磁偶极矩所导致.同时H—Be键在高覆盖度的极化也导致了静电能的减少.
2.2.2 多氢原子吸附Be(0001)表面
为了研究高覆盖度下多氢原子吸附Be(0001)表面的稳定问题,对可能的超原胞结构5H、p(3×3)/7H、p(3×3)/8H、p(3×3)/9H、p(3×3)/10H、 p(3×3)/11H、p(3×3)/12H、p(3×3)/13H、p(3×3)/14H进行了计算.首先结构优化得到多个氢原子稳定吸附Be(0001)表面的位置为:覆盖度低于0.67 ML(约6H/ 9Be),氢原子由于相互的排斥作用,不易于占据在近邻的位置,而是占据在fcc或hcp的位置.H—Be键长范围在0.146-0.160 nm之间.覆盖度为0.78 ML (约7H/9Be)时,fcc、hcp和桥位均能稳定占据氢原子.覆盖度在0.89-1.0 ML(约8H/9Be到9H/9Be)时,氢原子完全占据在桥的位置,H—Be键长约0.148 nm.这里可以得出,多个氢原子吸附Be(0001)表面的吸附位置随覆盖度的变化趋势与单个氢原子吸附Be(0001)表面的变化趋势相似.即低覆盖度时,氢原子易于占据在中空位置(hcp、fcc),高覆盖度时,氢原子容易吸附在近桥位.然而,当覆盖度大于1.0 ML时,氢原子只占据在hcp位和桥位.部分氢原子与最外层表面铍原子发生相对位移,如图2所示.H—Be键长约为0.142-0.155 nm.覆盖度在1.33(12H/9Be)时,氢原子吸附于Be(0001)表面到达饱和.更高覆盖度下,没有发现稳定的氢吸附的Be(0001)表面结构.
在本工作中,我们模拟了文献[19]中p(3×3)/12H结构,考虑了释放所有吸附原子坐标的初始结构,并与文献固定吸附原子坐标的情况进行了比较.结果发现,不同模型中氢原子吸附在Be(0001)表面的吸附能分别为-2.22 eV(自由氢原子),-2.01 eV(固定zH),和-1.91 eV(固定xH,yH,zH).实际上,Allouche报道的p(3×3)/12H超原胞结构相对于氢分子的结合能来说是不稳定的.他们得到的吸附能(约-2.0 eV,固定zH或xH,yH,zH)小于自由的氢原子在氢分子中的结合能(约-2.27 eV).氢原子在这个超原胞结构中更易于结合成分子而不是吸附在Be(0001)表面.因此,对此结构分析可知,文献[19]中所采用的模型以及氢原子吸附Be(0001)表面的计算结果是具有争议的.本文所有的结构优化计算将吸附原子的坐标完全释放(包括文献[19]固定的zH),有理由认为,我们的计算结果更接近氢吸附Be(0001)表面的物理本质.在我们的计算框架下,得到了一系列新的、高覆盖度下相对氢分子稳定存在的超原胞结构p(3×3)/10H、p(3× 3)/11H、p(3×3)/12H,如图2所示.发现氢原子吸附的垂直高度不同,但是它们平行于表面原子,排列成间距约为0.227 nm的线状结构.并且每个氢原子有2-3个最近邻铍原子,与文献[19]中每个氢原子有3个近邻铍原子的结构有所出入.我们期望,高覆盖度下(1.0-1.33 ML)原子氢线性排列于Be(0001)表面的吸附特性能在Be(0001)薄膜的相关材料中得到应用(例如储氢等物理应用).新结构中,氢原子与最近邻铍原子间的距离范围在0.142到0.155 nm之间(氢在桥位上的H—Be键长约0.142-0.145 nm,在hcp位上的H—Be键长约0.152-0.155 nm),与文献[19]中的结果(0.134-0.150 nm)也有所差别.如图2所示,新吸附结构中部分最外层铍原子在近邻氢原子的作用下发生剧烈膨胀,使得近邻氢原子渗入到铍表面次层.在能量上,图2(c)中p(3×3)/12H结构的吸附能约为-2.31 eV,比自由氢原子在氢分子中的结合能略高40 meV.与文献[19](图2(d))相比,新的吸氢结构(图2(c))较为稳定.图3给出了氢吸附Be(0001)表面的吸附能随氢原子覆盖度增加的变化趋势,其中虚线表示氢原子在氢分子中的结合能,同时文献[16,19]中相应吸附能的曲线也一并给出.需要指出的是,吸附能量上的差别是由于能量基准点选取不同所导致.曲线中在0.33和1.0 ML情况下得到氢吸附Be(0001)表面的相关性质(dH—Be、h、Φ)均与文献[16]结果吻合.在低覆盖度下(约0.2 ML),没有发现文献[19]报道的最小吸附值.图3显示,在我们的计算范围内氢原子在0.06-1.33 ML的结构中均能稳定地吸附于Be(0001)表面.与文献吸附能变化趋势不同的是,在0.65-0.78和0.89-1.0 ML区间均出现了平台.我们分析认为这种相变的驱动机制与氢原子的稳定吸附位有密切联系.在0.65-0.78 ML范围时,氢原子稳定占据在fcc或hcp的位置,而覆盖度为0.89-1.0 ML时,氢原子完全占据在桥的位置.其中0.78 ML成为拐点是由于fcc、hcp和桥位均稳定吸附了氢原子.在1.0 ML附近,吸附于Be(0001)表面的氢原子较于氢分子中的原子而言稍略为稳定.随后,更高覆盖度结构的吸附能在能量上出现了下降,最主要的原因是由于氢原子的吸附高度发生了明显的变化(分别占据在不同高度(见图2))导致氢与Be(0001)表面原子的相互作用加强.表现为Be表面原子发生膨胀以及近邻氢原子渗入到铍的次表面.此时Be衬底有相变发生,每个Be原子周围有2-3个最近邻氢原子,其中2个最近邻的氢原子与Be原子的H—Be键长约0.142-0.145 nm.由于Be单质能与氢生成金属氢化物BeH2,在氢原子高覆盖度下Be(0001)表面是否会有氢化物的形成呢?文献[34]报道了BeH2精确的实验数据,给出了气态BeH2的结构(线性)和H—Be键长(0.133 nm).如图2所示,在我们的工作中表面Be原子与近邻氢原子基本没有形成线性排列,并且近邻H—Be键长较于BeH2的键长略长约0.01 nm.因此,我们判断在高覆盖度下Be(0001)表面形成氢化物的可能性不大.图3中未给出大于1.33 ML结构的吸附能,因为吸附能均小于-2.27 eV.吸附表面的氢原子易于结合为氢分子,而不能稳定吸附Be(0001)表面.因此可以判断,当氢原子覆盖度大于1.33 ML时,氢原子吸附Be(0001)表面趋于饱和.通过偶极修正计算进一步证实(见图4),当覆盖度大于1.0 ML时,修正值明显低于其它低覆盖度的情况,表明氢原子数的增加减少了在Be(0001)表面的修正,饱和吸附原子在高覆盖度出现.

表2 不同覆盖度和吸附位上单个氢原子吸附在Be(0001)表面的相关性能Table 2 Properties of hydrogen adsorption on the Be(0001)surface for different coverages and adsorption sites
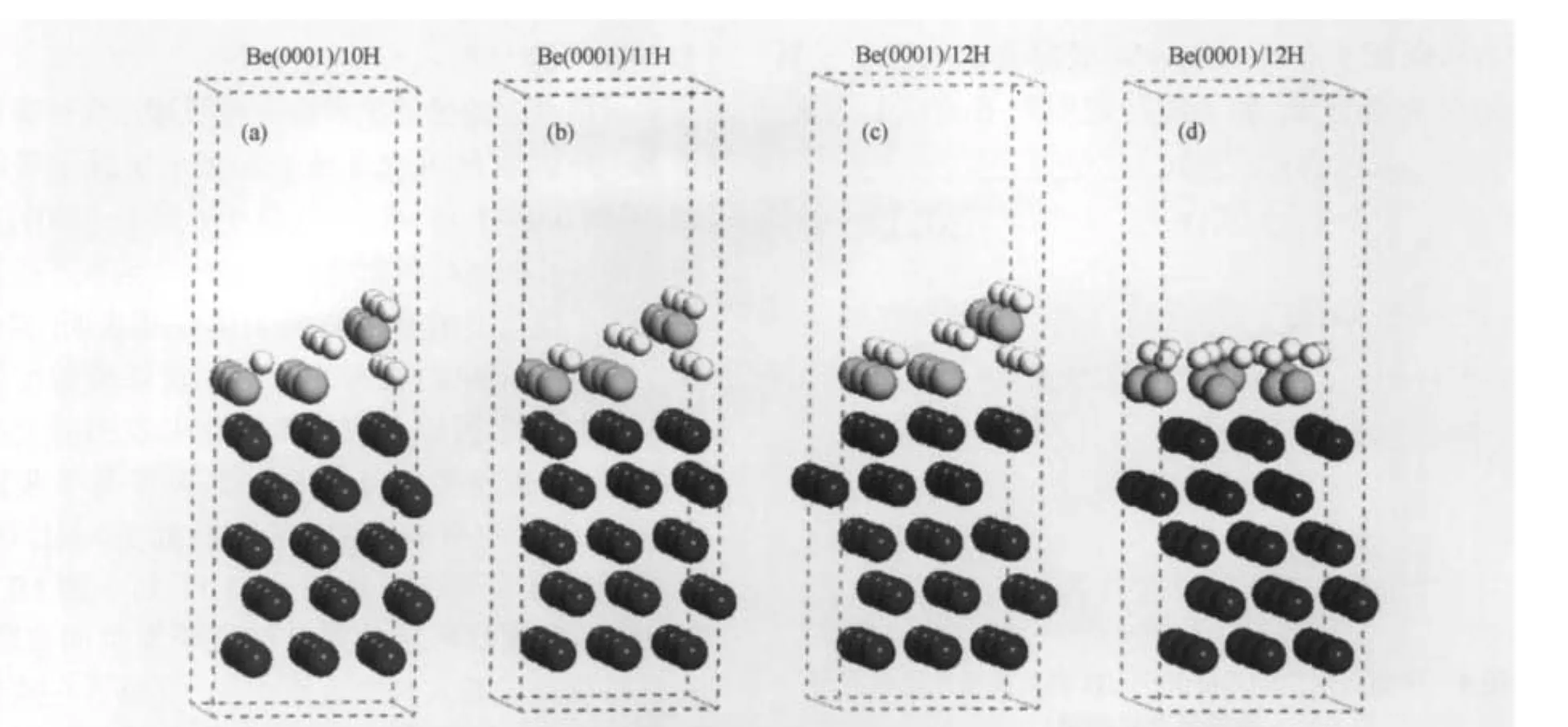
图2 Be(0001)/10H(a)、Be(0001)/11H(b)、Be(0001)/12H(c,d[19])结构的侧面图Fig.2 Side views of Be(0001)/10H(a),Be(0001)/11H(b),and Be(0001)/12H(c,d[19])structuresThe substrate beryllium atoms are in dark and the top-most beryllium atoms are in grey,while the hydrogen atoms are shown in white balls.
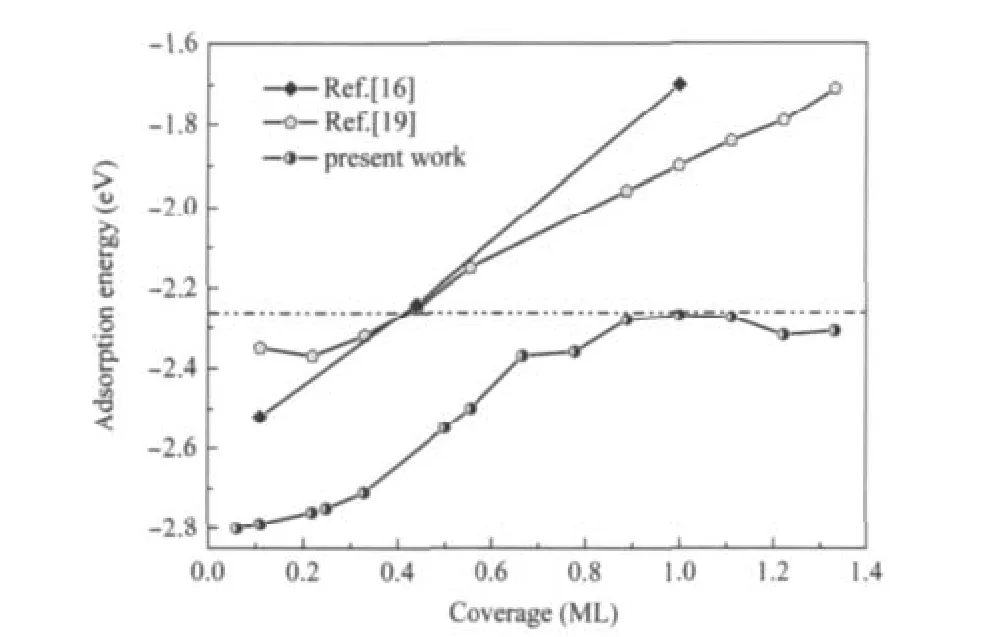
图3 以氢原子能量为基准计算的Be(0001)/H吸附能随氢原子覆盖度的变化趋势Fig.3 Calculated adsorption energy with respect to energy of atomic hydrogen on Be(0001)surface in theequilibrium adsorption sites versus coverage The value of half the theoretical binding energy(ca 2.27 eV)of H2molecule is represented by dashed line.
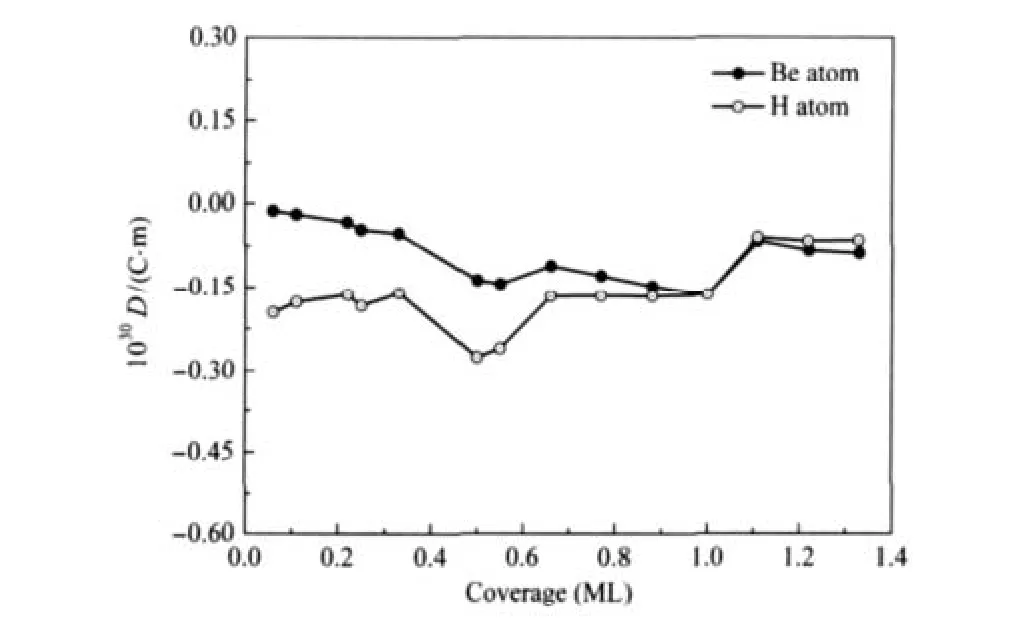
图4 平衡吸附位的Be(0001)/H表面偶极矩随氢原子覆盖度变化趋势Fig.4 Surface dipole moment of hydrogen adsorption on Be(0001)surface in the equilibrium adsorption sites versus coverage

图5 氢原子与最外层铍原子的平均垂直高度随氢原子覆盖度变化趋势Fig.5 Average vertical distances between hydrogen atoms and top-most substrate atoms versus coverage
最后,图5给出了氢原子相对最外层铍原子的平均垂直高度随覆盖度的变化关系.如图所示,覆盖度小于1.0 ML时,氢原子的平均垂直高度随着覆盖度的增加而增加.大于1.0 ML时,平均垂直高度明显减少.1.33 ML时为最小值(0.073 nm),达到饱和吸附垂直高度.
3 结论
采用密度泛函理论和第一性原理总能计算研究了Be(0001)表面单个氢原子和多个氢原子吸附问题.得到0.06-1.33 ML下氢原子吸附Be(0001)表面的结构、层间弛豫、吸附能以及表面功函数等性质随着氢原子覆盖度的变化关系.计算结果表明,多个氢原子吸附Be(0001)表面的吸附位置随覆盖度的变化趋势与单个氢原子吸附Be(0001)表面的变化趋势相似:覆盖度低于0.67 ML,氢原子易于吸附在fcc或是hcp中空的位置;在0.89到1.0 ML区间,氢原子最佳吸附位于近桥位;1.11到1.33 ML时, Be(0001)表面最外层的部分铍原子发生明显膨胀,其近邻氢原子渗入到铍表面次层.氢原子在能量上易占据在hcp位和桥位.相对于氢分子稳定存在的Be(0001)/10H、Be(0001)/11H、Be(0001)/12H吸附结构的提出还未见报道.当覆盖度超过1.33 ML,氢原子不能稳定吸附在Be(0001)表面.同时偶极修正和氢原子吸附垂直高度随覆盖度的变化分析可以判定:在1.33 ML氢吸附Be(0001)表面达到饱和.
1 Biswas,R.;Hamann,D.R.Phys.Rev.Lett.,1986,56:2291
2 Mattsson,T.R.;Wahnström,G.;Bengtsson,L.;Hammer,B.Phys. Rev.B,1997,56:2258
3 Bhatia,B.;Sholl,D.S.J.Chem.Phys.,2005,122:204707
4 Ledentu,V.;Dong,W.;Sautet,P.;Kresse,G.;Hafner,J.Phys.Rev. B,1998,57:12482
5 Dong,W.;Kresse,G.;Furthmüller,J.;Hafner,J.Phys.Rev.B, 1996,54:2157
6 Chou,M.Y.;Chelikowsky,J.R.Phys.Rev.B,1987,59:1737
7 Abramov,E.;Riehm,M.P.;Thompson,D.A.;Smelter,W.W. J.Nucl.Mater.,1990,175:9
8 Vajeeson,P.;Ravindran,P.;Kjekshus,A.;Fjellvag,H.Appl.Phys. Lett.,2004,84:34
9 Marino,M.M.;Ermler,W.C.;Tompa,G.S.;Seidl,M.Surf.Sci., 1989,208:189
10 Ray,K.B.;Hannon,J.B.;Plummer,E.W.Chem.Phys.Lett., 1990,171:469
11 Doerner,R.P.J.Nucl.Mater.,2007,363:32
12 Reinelt,M.;Linsmeier,C.Phys.Scr.,2007,128:111
13 Yu,R.;Lam,P.K.Phys.Rev.B,1989,39:5035
14 Hedin,L.;Lundqvist,B.I.J.Phys.C,1971,4:2064
15 Marino,M.M.;Ermler,W.C.J.Chem.Phys.,1991,94:8021
16 Stumpf,R.;Feibelman,P.J.Phys.Rev.B,1995,51:13748
17 Feibelman,P.J.Phys.Rev.B,1993,48:11270
18 Stumpf,R.Phys.Rev.B,1996,53:4253
19 Allouche,A.Phys.Rev.B,2008,78:085429
20 Kresse,G.;Furthermüller,J.Comput.Mater.Sci.,1996,6:15
21 Kresse,G.;Furthermüller,J.Phys.Rev.B,1996,55:11169
22 Vanderbilt,D.Phys.Rev.B,1990,41:7892
23 Blöchl,P.E.Phys.Rev.B,1994,50:17953
24 Perdew,J.P.;Burke,K.;Ernzerhorf,M.Phys.Rev.Lett.,1996,77: 3865
25 Perdew,J.P.;Burke,K.;Ernzerhorf,M.Phys.Rev.Lett.,1997,78: 1396
26 Monkhorst,H.J.;Pack,J.D.Phys.Rev.B,1976,13:5188
27 Payne,M.C.;Teter,M.O.;Allan,D.C.;Arias,T.A.; Joannopoulos,J.D.Rev.Mod.Phys.,1992,64:1045
28 Davis,H.;Hannon,J.;Ray,K.;Plummer,E.W.Phys.Rev.Lett., 1992,68:2632
29 Feibelman,P.J.Phys.Rev.B,1992,46:2532
30 Antonelli,A.;Khanana,S.N.;Jena,P.Surf.Sci.,1993,289:L614
31 Pohl,K.;Cho,J.H.;Terakura,K.;Scheffler,M.;Plummer,E.W. Phys.Rev.Lett.,1998,80:2853
32 Holzwarth,N.A.W.;Zeng,Y.Phys.Rev.B,1995,51:13653
33 Song,H.Z.;Zhang,P.;Zhao,X.G.Acta Phys.Sin.,2007,56(1): 465 [宋红州,张 平,赵宪庚.物理学报,2007,56(1):465]
34 Bernath,P.F.;Shayesteh,A.;Tereszchuk,K.;Colin,R.Science, 2002,297:132
January 14,2010;Revised:March 12,2010;Published on Web:June 28,2010.
Density Functional Theory Study on Hydrogen Adsorption on Be(0001)Surface
NING Hua TAO Xiang-Ming WANG Mang-Mang CAI Jian-Qiu TAN Ming-Qiu*
(Department of Physics,Zhejiang University,Hangzhou 310027,P.R.China)
We report on density functional theory(DFT)total-energy calculations within the generalized gradient approximation for the adsorption of hydrogen onto Be(0001)surface.To investigate the atomic geometries and stability with different hydrogen coverages for this system,we changed the atomic hydrogen coverage from 0.06 to 1.33 monolayer(ML)using various surface supercell geometries.The calculations showed that the adsorption sites have a strong dependence on hydrogen coverage.The adsorbates mainly occupied fcc and hcp hollow sites below 0.67 ML. At 0.78 ML the hydrogen atoms were adsorbed on hollow and bridge sites while for the higher coverage range(ca 0.89-1.00 ML)the hydrogen atoms were adsorbed onto the tilted bridge sites,i.e.,a bridge site with a small deviation towards the hollow position.From 1.11 to 1.33 ML,the adsorbed hydrogen atoms were located at hcp and bridge sites, and some Be surface atoms were expanded.All these adsorption configurations were found to be energetically favorable with a H2reference point fixed on H2molecule.Further total-energy calculations based on a p(3×3)geometry did not revealed any stable or energetically favorable adsorption geometry versus the H2molecule beyond a hydrogen coverage of 1.33 ML.
Density functional theory;Be(0001)surface;Hydrogen adsorption;Adsorption energy
[Article] www.whxb.pku.edu.cn
*Corresponding author.Email:mqtan@zju.edu.cn;Tel/Fax:+86-571-87951328.
The project was supported by the Research Project of Department of Education of Zhejiang Province,China(Y200804278)and Program for Changjiang Scholars and Innovative Research Team of the Ministry of Education of China(IRT0754).
浙江省教育厅科研项目(Y200804278)和长江学者和创新团队发展计划(IRT0754)资助
O641;O647
