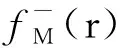双核镉配聚体及其衍生物荧光光谱发光机理的计算化学研究
2010-11-26刘文龙钟爱国刘述斌
黄 莺 ,刘文龙 *,钟爱国 , 刘述斌
(1.湖南中医药大学药学院, 中国 长沙 410208;2.台州学院化学系, 中国 临海 317000;3.Research Computing Center, University of North Carolina, Chapel Hill, North Carolina 27599-3420, USA;4.湖南师范大学化学化工学院, 中国 长沙 410081)
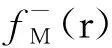
1 计算模型和方法
为获得标题物的分子结构和能量信息,运用 Gaussian 03 W 量子化学程序包,以 1 ([Cd2Cl4(Hbm)2],C16H18Cd2Cl4N4O3) 的 Cif 晶体结构文件为初始结构,采用 B3LYP/6-311+G(d) 方法优化所得构型为其基态构型, 见图 1.用 HF-CIS/6-311G(d) 优化其激发态 (S1) 构型, 用TD-DFT B3LYP/6-311+G(d) 计算 1 的发射光谱; 根据 B3LYP/6-311+G(d) 方法计算阳(阴)离子与其中性分子总能量之差得到第一电离势(I)和电子亲和势(A)用来计算密度泛函活性理论中的各种活性指数.
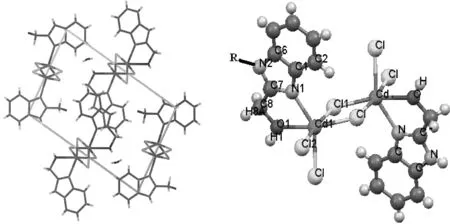
图1 镉配聚物(左)和模型分子 1 (右) 的结构(R= H; CH3; NH2; CN)
在密度泛函活性理论(DFRT)框架中,化学势μ、化学硬度η,被定义
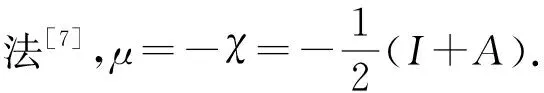
2 计算结果与讨论
2.1 标题物1的稳定几何构型
用DFT 在B3LYP/6-311+G(d)水平上, 对模型分子1进行优化计算, 得到的部分几何结构参数列于表1,比较计算结果和晶体学参数,发现键长最大相差 0.007 nm, 键角最大相差 0.4°,可见两者结果十分接近.含Cd 的五元螯合环与咪唑环接近于共平面,振动分析表明无虚频,因此采用该计算方法得到的分子几何构型是可靠的.
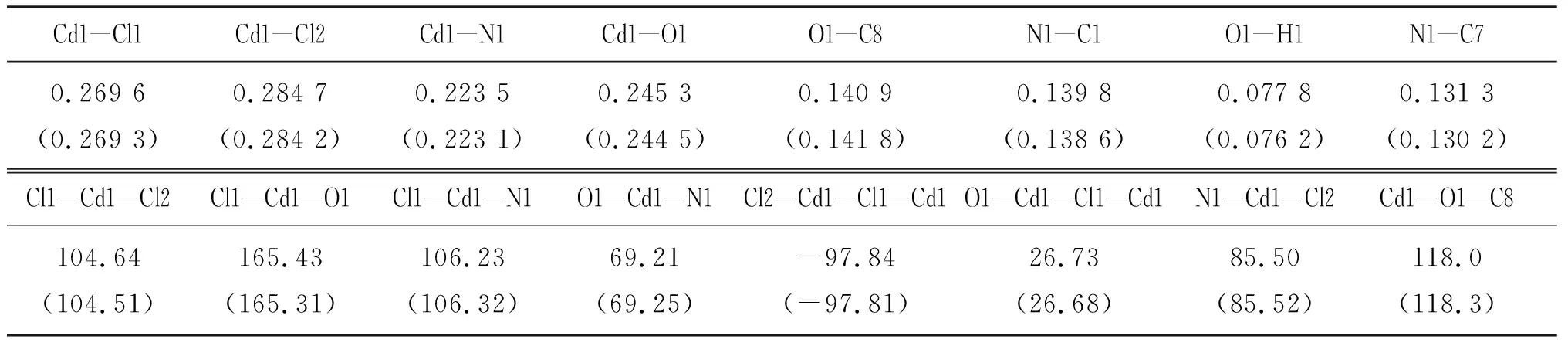
表1 一些选择的键长(单位:nm)和键角(单位:(°))的计算值与实验值比较
注:括号内数据为计算模拟值.
2.2 吸收光谱及前线分子轨道
金属有机化合物发光波长和发光效率取决于电子跃迁和能量转移机理.配合物吸收光谱与前线分子轨道关系密切.以标题物晶体结构作为分子模型, 我们探讨了不同泛函(基组)及溶剂对标题物紫外吸收光谱峰强度(f)和波长(λ)的影响,结果发现用 TD-DFT在 B3LYP/6-311+G(d) 水平上对1的基态分子进行的垂直跃迁模拟,比较接近于实验所测值(溶剂为水),具体模拟结果如图2所示.
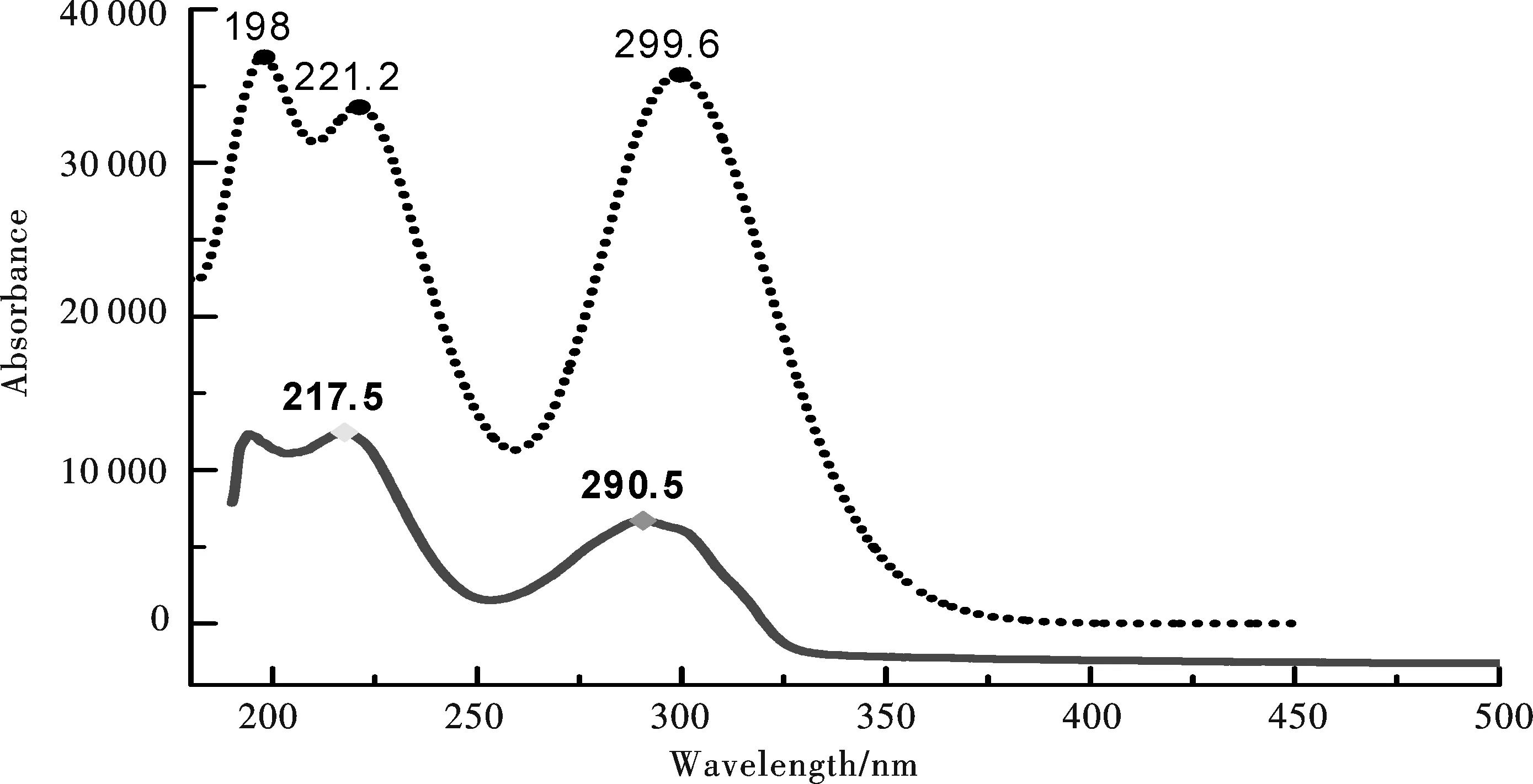
图2 标题物1的模拟电子吸收光谱(虚线为模拟值,实线为实验值)
用 B3LYP/6-311+G(d) 方法对 1 分子轨道成分进行自然键轨道分析, 用组分原子或分子片的轨道系数的平方和的百分比来表示该部分对某个分子轨道中的贡献, 将 1 所有原子分为 7 个部分: (1) 2Cd; (2) 2O; (3) 4N; (4) 4Cl; (5) 苯环 C(I); (6) 咪唑环 C(II); (7) 鳌合环 C(Ⅲ).同时也对类似物 ([M2Cl4(Hbm)2], M=Zn; Hg) 中金属原子的贡献进行了分析.计算结果如表 2 所示.

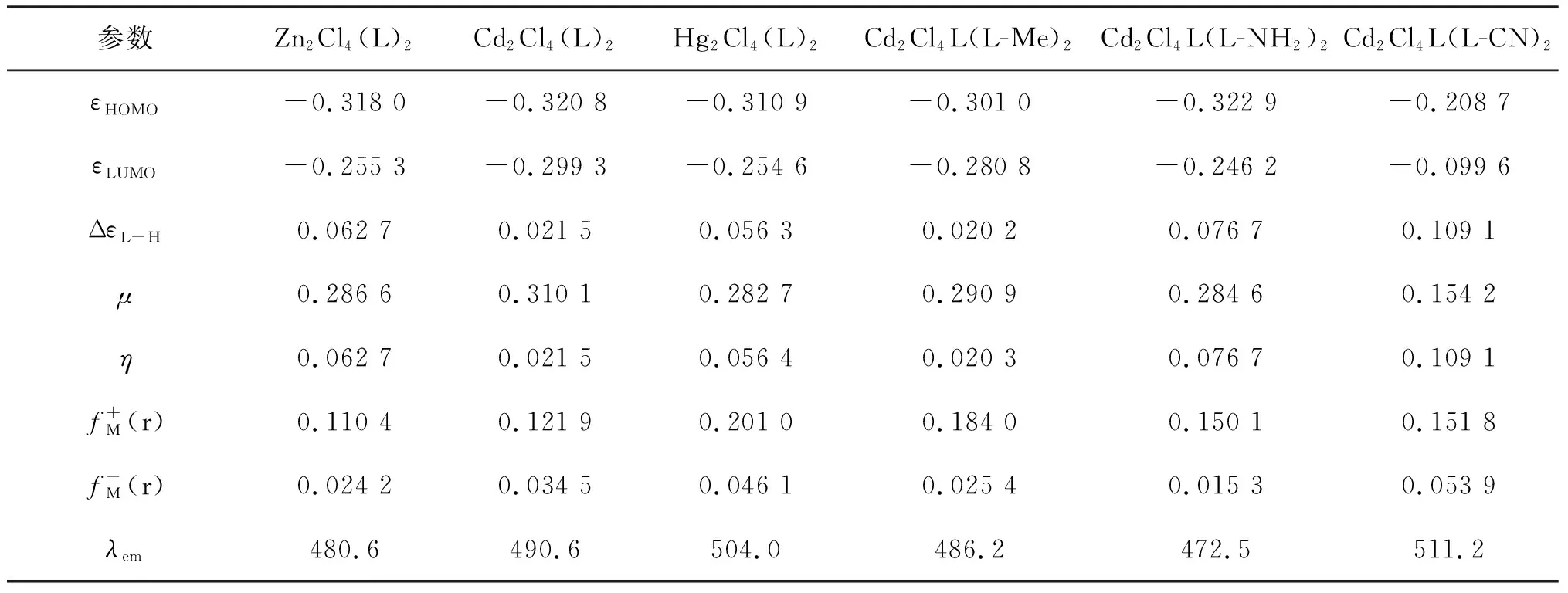
表2 标题物1及其衍生物前线分子轨道成分分析 %
结果表明,在最高占据分子轨道HOMO上,苯环C(Ⅰ)的贡献占5.3%,而咪唑环C(Ⅱ)和螯合环C(Ⅲ)也分别只占到2.1%,3.4%,其中4N占1.9%,2O占12.1%,基态分子的HOMO电子云集中分布在配体Cl上,占到73.5%.在最低空轨道LUMO上,苯环C(I)的贡献仅占17.3%,咪唑环C(II)占4.5%,其中4N占11.7%,而配体Cl只占到2.1%,金属Cd却占50.0%,说明激发态时LUMO电子云集中布居在金属离子Cd上.由此可以推知,电子从基态跃迁到第一激发态时,电子将在配体Cl和金属Cd之间进行转移.用晶体场理论描述的分子轨道分裂情况也证实了这一趋势(见图3).在八面体配体场作用下,金属中心离子M的5个简并d轨道劈裂为三重简并的t2g轨道和二重简并的eg轨道.配体Cl有充满电子的p-π轨道,其能级比中心离子的t2g轨道能量要低.按照双原子分子轨道原则:2个不同能级的原子轨道组成分子轨道时,成键分子轨道中将含有较多成分的低能级原子轨道,而反键分子轨道中则含有较多成分的高能级原子轨道.由此可见,配合物分子的成键π分子轨道,即配合物分子的最高占据轨道(HOMO)的成分接近配体的p-π轨道,而配合物分子的反键π分子轨道,即配合物分子的最低未占据轨道(LUMO)的成分较接近中心离子的t2g轨道.当配合物1被激发时,电子由HOMO跃迁到LUMO,即配体到金属的电荷跃迁[4].用Zn(Ⅱ)、Hg(Ⅱ)等过渡金属取代Cd(Ⅱ)的计算模拟结果,也支持了中心金属M在电荷转移-发光过程中的电子受体作用.图4为模型分子1的前沿分子轨道分布图,更加形象地表明了前线轨道的组成特征.

图3 标题物1的可能发光机理
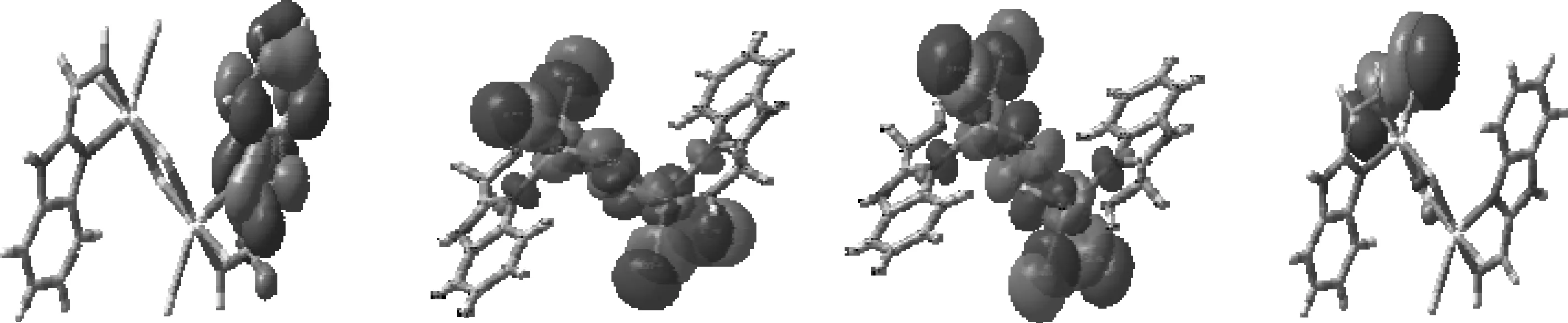
HOMO-1 HOMO LUMO LUMO+1图4 标题物1的前线分子轨道
2.3 电子亲和能和发射光谱
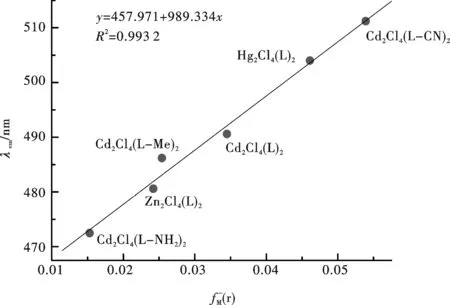
用CIS/6-311+G(d)优化1及其5种衍生物的最低激发单重态S1的几何结构,然后用TD-DFT在B3LYP/6-311+G(d)水平下计算出它们的电子亲和势(A)和发射光谱波长,结果见表3.从表3中可以看出,除[Cd2Cl4(Hbm)(Hbm-CN)]的电子亲和势较大外(1.07 eV)(它可能具有更好的电子传输性能),其余5种衍生物的电子亲和势相近;1的发射光谱的计算结果(气相λcal.,max=490 nm)与实验值(在甲醇中测得最大吸收峰位λexp.,max= 480.6 nm;固体中为 500 nm) 十分接近.计算结果还表明, 随着金属M原子序数增加(Zn 表3 Cd2Cl4(L)2及其衍生物电子亲和势和发射光谱波长的模拟结果 注:L=Hmb; A 的值属 TD-DFT 计算值;1的实验值λexp.,max= 480.6 nm(气);λexp.,max=500 nm(固). 表4 标题物1及其衍生物的DFT指数和发射光谱波长 单位:nm 注:L = Hmb; 标题物 1 的实验发射值为 490 nm. 图5 金属离子的亲电福井函数和发射波长λem 的关系(注:L=Hmb) 参考文献: [1] 陈丽华,王展旭,林希伟. 镉(Ⅱ)与对苯二甲酸和2,2′-联吡啶配合物光谱性质的理论研究[J]. 分子科学学报,2007,23(2):143-145. [2] 樊文浩,郝玉英,房晓红,等.取代基对8-羟基喹啉锂电子光谱影响的理论研究[J]. 发光学报,2007,28(2):193-197. [3] 李小兵,王学业,禹新良,等. 8-基喹啉阴离子的锌配合物及其衍生物的电子光谱性质的含时密度泛函理论研究[J]. 化学学报, 2006,64(3):208-212. [4] 刘晓冬. 发光金属配合物中的吸推电子效应对电子能级结构和电子光谱影响的理论研究[D]. 吉林:吉林大学,2006. [5] 李巧云, 马运声,杨高文,等. 超分子2,2-联咪唑镉(Ⅱ)配聚物的合成与结构表征[J]. 无机化学学报,2008,24(9):1 461-1 467. [6] 王锡森,黄雪峰,熊仁根. 5-取代 1-氢四唑合成中捕获的一个新奇的三维镉配聚物[J]. 无机化学学报,2005,21(7):1 020-1 024. [7] 刘光祥,陈 宏,任小明. 二维层状镉配合物[ CdCl2(obbm) ] 的合成、晶体结构及荧光性质[J]. 无机化学学报, 2010, 26(1):161-165. [8] PEARSON R G. Hard and soft acids and bases[J]. J Am Chen Soc, 1963,85(22):3 533-3 539. [9] KOOPMANS T. Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektonen eines atoms[J]. Physica, 1933, 1:104-113. [10] PARR R G, YANG W T. Density functional approach to the frontier-electron theory of chemical reactivity[J]. J Am Chem Soc, 1984, 106:4 049-4 050. [11] 刘述斌.概念密度泛函理论及近来的一些进展[J].物理化学学报, 2009, 25(3): 590-600.
2.4 DFRT 指数和发射光谱波长关系
3 结论