介孔氧化铝及其前体的晶体结构与表面化学的研究进展
2010-11-09关丽丽柳云骐李广慈刘晨光
关丽丽,柳云骐,李广慈,刘晨光
(中国石油大学 (华东)重质油加工国家实验室 CNPC催化重点实验室,山东 青岛 266555)
进展与述评
介孔氧化铝及其前体的晶体结构与表面化学的研究进展
关丽丽,柳云骐,李广慈,刘晨光
(中国石油大学 (华东)重质油加工国家实验室 CNPC催化重点实验室,山东 青岛 266555)
综述了介孔氧化铝及其前体的晶体结构与表面化学的研究进展。从介孔氧化铝的主要合成路线出发,对介孔氧化铝的控制合成进行了系统总结。并对近几年合成的新型氧化铝(包括规整孔道介孔氧化铝、不同形貌氧化铝、纳米晶氧化铝组装体等)进行了详细介绍。重点介绍了介孔氧化铝及其前体的晶体生长及控制策略、铝的微环境和表面羟基结构等方面的研究。对该领域的热点问题和存在的研究机遇进行了展望,提出要加强对氧化铝的介观层面的研究。
介孔氧化铝;介孔氧化铝前体;晶体结构;表面化学
仅从氧化铝的分子式 A12O3而言,它似乎是一种很简单的氧化物,但当考虑其晶体结构及空间因素时发现,它是一种形态变化复杂的两性化合物。在已知的 9种晶型 (χ-,β-,γ-,δ-,κ-,θ-,ρ-,η-,α-A12O3)中,催化剂或载体最常用的氧化铝多为活性氧化铝 (即γ-A12O3和η-A12O3),这主要是由氧化铝及其前体的性质决定的。由于不同晶型之间,即使是同一种晶型,它的宏观及微观结构性质 (如密度、孔体积、孔分布、比表面积等)也会依其制备方法不同而有很大差异。这种结构变化的多样性,一方面决定了它的应用广泛性,另一方面又为掌握其规律性带来了困难。所以,有关活性氧化铝的晶体结构及其表面化学规律,迄今仍是十分活跃的研究领域。
对氧化铝及其前体的晶体结构及其表面化学规律的认识,对于选择不同的起始原料和调节活性氧化铝的制备路线及其工艺参数,得到满足不同要求的活性氧化铝产品具有重要的意义。随着合成化学和现代分析技术的发展,活性氧化铝的新品种不断出现,极大地丰富了活性氧化铝及其前体的晶体结构及其表面化学的研究。
本文对介孔氧化铝的晶体结构及其表面化学规律的最新研究进展进行了系统的综述,并对关注的热点问题和存在的研究机遇进行了展望。
1 介孔氧化铝的控制合成
介孔氧化铝的控制合成方法有多种,常用的合成方法有:沉淀法、水热或溶剂热及表面活性剂辅助水热或溶剂热法、溶胶 -凝胶法、溶胶 -水热法等。不同的合成方法,即使相同的合成方法中制备参数的调变都会引起所合成的介孔氧化铝形貌及物性的改变。近年来除了研究创新合成路线及其过程参数对介孔氧化铝理化性质的影响外,纳米介孔氧化铝及其介观层面的形貌组装的研究也受到关注,一维的纳米棒、纳米管、纳米线、纳米片等以及组装体 (如三维花状的纳米氧化铝)都已相继成功合成。
1.1 一维纳米晶氧化铝
介孔氧化铝一般由沉淀法得到的具有不同粒度和不同结晶度的拟薄水铝石经高温焙烧得到,由于前体的颗粒大小和结晶状态对介孔氧化铝晶体形成的影响较大,而且高温焙烧过程中氧化铝晶粒的团聚明显,因此这种方法很难得到真正意义上的纳米晶,近年来发展了水热或溶剂热法合成氧化铝纳米晶的技术,该方法制备的氧化铝形貌较其他方法更具可控性,因此目前对采用该方法合成不同形貌氧化铝纳米晶的研究较多。晶体介观形貌的形成是源于晶面的定向生长,可通过杂质离子的选择性吸附或添加剂在已有的结构上取向附生而实现[1]。由于添加剂会不可避免地在制备过程中引入杂质,限制了所制备氧化铝的应用,因此相对于添加剂控制的晶体生长,通过制备原料的选取及过程参数的调变来合成不同形貌的介孔氧化铝更有实际意义。
Yang等[2,3]以 HgC l为介质、铝片为原料,在室温下合成出纤维状的无定形纳米氧化铝前体,直径为 5~15nm,长度可达几微米,850℃下焙烧 2h后变为纤维状纳米晶γ-A l2O3。为进一步了解纤维状纳米氧化铝的形成过程,他们缩短了铝片的浸渍时间,降低了 HgCl溶液的浓度,发现铝片表面有垂直生长的钟乳石状产物出现,当将这些定向生长的产物移去之后发现铝片表面有很多小洞,初步猜测纤维状的纳米氧化铝就是从这些洞里生长出来的,并且这些小洞占铝片孔体积的比例很大。以 HgC l为介质可以使铝原子在汞齐/空气界面层与空气中的氧气和水反应,氧化铝在界面间定向自组装得到氧化铝纤维;并且所用铝片的纯度越高,制得的氧化铝比表面积越大。张立岩等[4]采用 N aA lO2-CO2法也合成了γ-A l2O3,并发现当煅烧温度从500℃升至 1 200℃时,氧化铝粒子的形貌由纤维状变为球形。
在无任何添加剂的情况下,仅通过改变铝源的种类也会对最终产品的形貌及物性产生一定的影响。Zhang等[5]在不添加任何修饰剂的情况下,以A lC l3·6H2O为铝源,将 N aOH溶液滴加到 A lC l3溶液中,调节溶液 pH至 4.0,在 200℃的高压釜中,当反应时间延长到 24h时得到直径为 60nm、长约几微米的纤维状纳米氧化铝。M ishra等[6]分别以A l2(SO4)3,A lCl3,A l(NO3)3为原料合成薄水铝石,发现以 A l2(SO4)3为原料合成的薄水铝石比以A lC l3和 A l(NO3)3为原料合成的薄水铝石的结晶水含量低、堆密度小、比表面积大。TG-D TA表征结果显示,薄水铝石转变为γ-A l2O3的吸热峰在460~480℃之间,并且温度升高到 1 000℃没有发生相变,表明用水热方法得到的γ-A l2O3具有良好的热稳定性。Guzm an-Castillo等[7]也考察了分别以A l2(SO4)3和 A lCl3为铝源对最终产物的影响,发现用A lC l3合成的薄水铝石比用 A l2(SO4)3合成的薄水铝石结晶度高,并且经 600℃焙烧转变为γ-A l2O3后只具有 L酸活性;而以 A l2(SO4)3为原料得到的γ-A l2O3既具有 L酸活性又具有 B酸活性。说明铝源种类的不同会对最终产物的物性产生影响。
另外,有关介质酸碱度对产物形貌的控制问题,Chen等[8,9]用 A l(NO3)3·9H2O和乙二胺为原料,通过调变溶液 pH,用水热法研究了棒状和片状纳米氧化铝的控制合成。当 pH=5时,所得γ-A lOOH为棒状 (见图 1A),棒长为 100~150nm,平均宽度为 8nm,棒沿 (001)晶面生长。在 600℃下焙烧 3h后,得到大量的棒状γ-A l2O3,且γ-A l2O3纳米棒沿 (111)晶面生长 (见图 1B)。并且因为失水,所得棒长变短,空隙率增大。而当 pH=10时,得到长为 20~30nm的片状γ-A lOOH。经观察发现,在γ-A lOOH转变为γ-A l2O3的过程中,γ-A lOOH的 (010)晶面的层间氢键消去了,这可能是导致晶体结构和化学组成变化的原因。

图 1 pH=5时γ-A lOOH(A)和γ-A l2O3(B)纳米棒的 TEM图像[9]Fig.1 TEM images ofγ-A lOOH(A)andγ-A l2O3(B)nano-rods prepared at pH=5[9].
1.2 规整孔道介孔氧化铝
具有规整孔道的介孔氧化铝因其孔分布较窄、比表面积大,表面不同的电势使金属离子更易负载,在催化领域中具有重要的应用价值[10]。也有研究者认为,载体的孔道是否规整有序对催化剂的活性没有影响[11]。但如果能合成具有规整孔道的氧化铝材料,将为后续的其他研究奠定基础。现在能一步合成孔道规整的介孔氧化铝的报道并不多,而且对具有三维连通孔道的立方相介孔氧化铝的报道更少。
目前合成具有规整孔道介孔氧化铝的方法主要基于表面活性剂胶束的软模板机理,合成出的介孔氧化铝多为蠕虫状孔道,表面活性剂在合成中仅起到控制孔径的作用。1996年,Bagshaw等[12]以电中性非离子表面活性剂为模板剂,以烷氧基铝为铝源,首次合成了具有蠕虫状织孔结构、比表面积达500m2/g的介孔氧化铝。此后相继出现了用不同模板剂合成介孔氧化铝的研究报道[13~18]。根据模板剂不同,具有分子筛性质的孔道规整的介孔氧化铝的合成方法及部分物性参数见表 1。
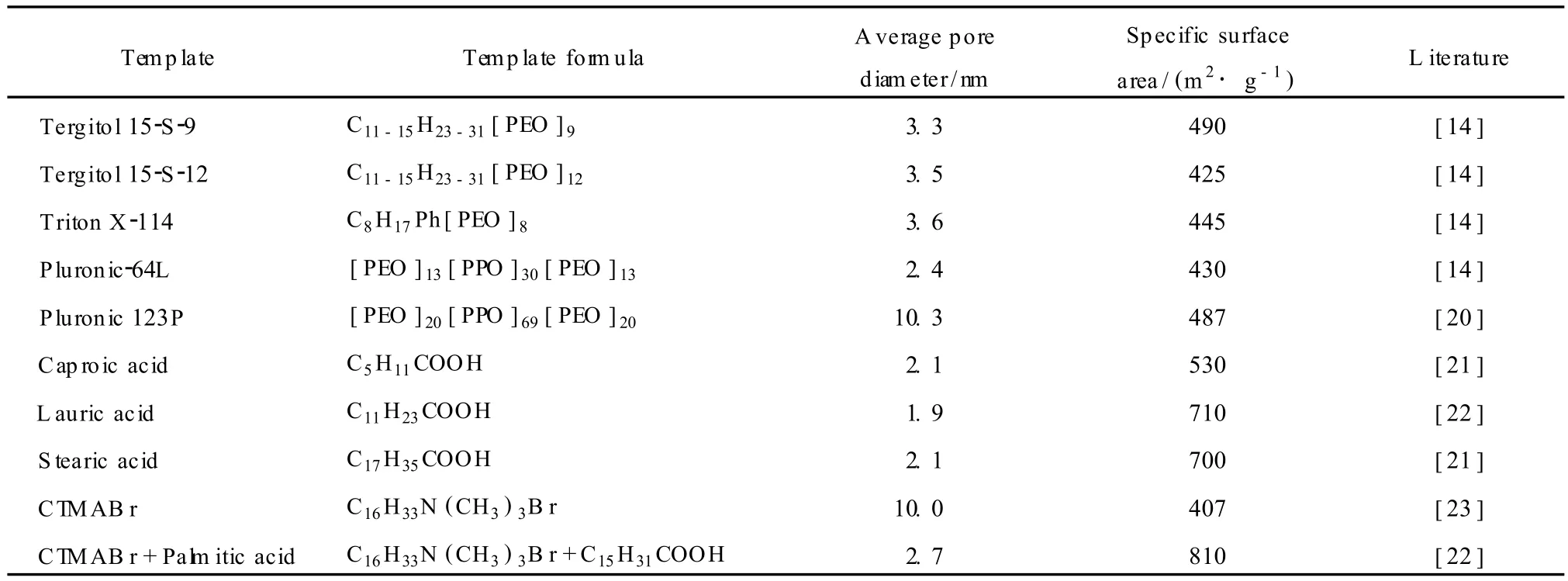
表 1 不同模板剂合成的的介孔氧化铝的平均孔径和比表面积[19]Table1 Average pore size and specific surface area of mesoporous alum ina prepared w ith different templates[19]
最近,李志平等[24,25]采用溶胶 -凝胶法,以异丙醇铝为铝源、异丙醇为溶剂、非离子表面活性剂Triton X-100和三嵌段共聚物 P123为复合模板剂,研制出比表面积大于 500m2/g、孔分布窄 (2~12nm)、孔体积大于 1.0cm3/g、平均孔径为5.7nm、孔道成蠕虫状且具有一定有序性的介孔氧化铝。
He等[26]以硫酸为形貌导向剂,以 A lCl3和NH4OH为原料,首先在 240℃下水热合成γ-A lOOH纳米棒,通过调节 H2SO4与 A l(OH)3的摩尔比 (从 0.012 5增加到 0.05),得到长 0.25~1.00μm、直径 40~10nm的纳米棒,然后经高温焙烧得到介孔氧化铝。另外,在不添加 H2SO4的情况下生成的γ-A lOOH为板状,的存在抑制了(020)晶面的生长,使晶体沿 (100)晶面生长。
Gan等[27]以异丙醇铝为原料,通过选用不同的反应溶剂,用溶剂热法合成了形貌可控的纳米结构介孔氧化铝。当以甲苯和异丙醇 (体积比 1∶1)的混合物为反应溶剂时,合成出长 50~80nm、外径8~10nm、内径 4~5nm、平均孔径 6.5nm、比表面积 365m2/g、孔体积 0.84cm3/g的管状薄水铝石纳米晶。当以去离子水和异丙醇 (体积比 1∶1)的混合物为反应溶剂时所得薄水铝石为多孔的片状结构,平均孔径为 10.8nm,但比表面积较小 (121m2/g),孔体积也不大于0.41cm3/g。而同样条件下仅用异丙醇为反应溶剂时,可得到薄水铝石纳米纤维结构,比表面剂达312m2/g,孔体积为 0.52cm3/g,平均孔径为5.4nm。还发现仅用甲苯为反应溶剂时,合成出的氧化铝为斜方晶形,并呈现由蠕虫状的薄水铝石或氧化铝组成的孔结构。但关于溶剂甲苯、异丙醇等如何影响介孔氧化铝均一孔的形成,目前仍在探索之中。
1.3 纳米晶氧化铝的组装体
随着一维纳米晶氧化铝的成功合成,由一维纳米晶氧化铝组装成各种形貌的三维纳米聚合体成为可能。L iu等[28]以乙醇为反应溶剂,合成出长约9μm、宽 4.5μm、厚 60~90nm的叶片状薄水铝石(见图 2A)。因为乙醇的存在一方面抑制了前体的快速水解,另一方面在水中形成氢键,强化了定向生长机制。进一步研究发现,在晶化过程中,当乙醇的浓度增加到 0.8m ol/L时,采用叶片自组装会形成直径约为 10μm的花状结构,经 600℃焙烧后得到γ-A l2O3,且形貌仍为花状 (见图 2B)。

图 2 纳米片状 (A)[28]、三维花状 (B)[28]、玫瑰花状 (C)[29]和海胆状 (D)[30]结构的 SEM图像Fig.2 SEM images of boehm ite nanoleaves(A)[28],3-D flower-like(B)[28],rose-like(C)[29]and urchin-like(D)[30]morphologies.
关于花状薄水铝石结构的组装,M azloum i等[29]曾做过较为详细的研究。由于氢键作用,一维花瓣自组装形成了三维玫瑰花状结构 (见图 2C)。不仅是花状的,而且形状更为奇特的三维纳米氧化铝也已被研制出来。W u等[30]选用 A l2(SO4)3和尿素为原料,以四氢呋喃为溶剂,合成出壁厚为300~800nm、孔径约为 4μm的中空海胆状的薄水铝石结构。并且发现添加浓度为 0.068 3m ol/L的尿素时可得到自封闭的γ-A lOOH微球,而尿素浓度增加到 0.095 4m ol/L时可得中空海胆状的薄水铝石结构。在 800℃下焙烧 4h转变为γ-A l2O3,形貌仍保持不变 (见图 2D)。如果其他一些有用的颗粒可以被包覆在壳中,那么这种自封闭的结构体系将在纳米科学和纳米科技领域发挥巨大的作用。
2 介孔氧化铝及其前体的晶体结构
2.1 拟薄水铝石和薄水铝石的晶体结构
薄水铝石属斜方晶系,为层状结构,层与层之间靠氢键连接在一起,铝离子处于扭曲的氧八面体中,如图 3所示。拟薄水铝石具有相似的结构,但含过量水,每个 A l2O3分子含有 1.5~2.5个 H2O分子,结晶度差。Guzm an-Castillo等[31]通过 XRD和一些改性的技术对拟薄水铝石和薄水铝石的晶体结构进行了分析,发现虽然它们的晶体结构相同,晶格参数和晶胞中的原子位置相似,但是晶粒尺寸不同,拟薄水铝石的晶粒较小。进一步研究发现铝与氧原子间的键长决定了晶胞大小,从而造成晶面垂直于 (100),(010),(001)等不同方向,对于大晶粒,垂直于 (010)方向的晶面对晶粒表面特性影响最大;而对于小晶粒,垂直于 (100)和 (001)方向的晶面贡献最大。氢氧化铝在加热过程中发生吸热的脱水反应,利用该过程差热曲线中吸热峰的温度可以鉴定出不同晶相,如薄水铝石和拟薄水铝石分别在 500℃及 400~500℃处出现吸热峰;另外,根据 XRD谱图中特征单晶面间距 (d)及衍射峰的相对强度也能区别薄水铝石和拟薄水铝石[32],如d(020)<0.62nm时为薄水铝石,d(020)=0.66~0.67nm时为拟薄水铝石,d(020)=0.62~0.65nm时为薄水铝石和拟薄水铝石的中间产物。

图 3 γ-A lOOH的晶体结构[33]Fig.3 Structure ofγ-A lOOH crystal[33].
Hong等[34]采用 XRD,SEM,TEM技术来区分拟薄水铝石和薄水铝石,得出如下结论:薄水铝石为晶体,晶形完整,XRD衍射峰的相对强度高,而拟薄水铝石的结晶度差,晶形不完整,XRD衍射峰的相对强度低;SEM观测表明拟薄水铝石和薄水铝石分别为粒径 30μm和 5μm的球状聚集物;TEM观测表明拟薄水铝石初始粒子为起褶皱的片,而薄水铝石为菱形的晶体,最大粒径达到 50nm。
2.2 γ-A12O3和η-A12O3的晶体结构
氧化铝是氢氧化铝的脱水产物,由于氧原子和铝原子的空间堆叠方式及含水量不同,各种氢氧化铝经热分解形成一系列同质异晶体[35]。这些同质异晶体,有些呈分散相,有些呈过渡态,通常可以用XRD方法鉴定各种氧化铝及其水合物的晶相结构。在氧化铝各晶型的鉴定中,γ-A12O3和η-A12O3的鉴定是最困难的。
γ-A12O3和η-A12O3均为活性氧化铝,且其XRD衍射峰的位置、形状都相近[36]。γ-A12O3属于四角晶系,η-A12O3属于立方晶系,这两种氧化铝的晶格与尖晶石 (M gA l2O4)的结构很类似。尖晶石的单位晶胞是由 32个立方密堆积的氧原子和16个在 1/2八面体空隙中的铝原子以及在四面体空隙中的 8个镁原子构成。而在γ-A12O3中,只有21个铝原子分布在 24个阳离子部位,在八面体位置上有 2个空位,而 8个铝原子分布在四面体空隙中,相当于形式,其中“□”表示空位。γ-A12O3及η-A12O3的 XRD谱图上存在明显的弥散线条,这表示晶格很无序。对于γ-A12O3,这种无序性主要是由铝原子 (特别是处于四面体空隙中的铝原子)的无序性所决定的。而Paglia等[37]认为γ-A12O3的晶体结构有两种,包括四角晶系和立方晶系,低温时主要为四角晶系,而温度达到 750℃以上则主要为立方晶系。
2.3 介孔氧化铝及其前体的晶体缺陷结构
郝保红等[38]认为,实际晶体中并不是所有原子都严格按照周期性规律排列的,因为晶体中存在着一些微小的区域,在这些区域内或穿过这些区域时,原子排列的周期性将受到破坏,这样的区域称为晶体缺陷。晶体中存在的电子和空穴、杂质原子、键的变形以及晶体生长时的取向情况等,都是构成晶体不完整性的因素,而从催化作用来看,晶体中存在的各种缺陷与催化活性有一定的关联性,可以通过改变其生长条件来控制其内部缺陷的形成,从而改善和提高其质量和性能。介孔氧化铝及其前体的晶体缺陷结构早就引起了人们的注意。
Tertian等[39]认为薄水铝石脱水的速率决定其制得的γ-A12O3的结构。他们认为慢速加热和低的脱水速率会造成所制得的γ-A12O3晶格畸变,而快速加热脱水会产生有序的立方尖晶石γ-A12O3。Yanagida等[40]采用 XRD表征发现,在室温原位加热时γ-A12O3晶格畸变会减少。随后W ilson等[41]又研究了加热温度对γ-A12O3缺陷结构的影响,发现延长加热时间也会使γ-A12O3晶格畸变减少。Tsuchida等[42]发现若薄水铝石的结晶度高会引起产物的晶格畸变,明显的标识是在立方 (400)线有显著的分裂峰,并且所得γ-A12O3的结晶度也高;若前体的结晶度低,立方 (400)线的分裂峰会变得模糊不清。他们认为晶格畸变与加热过程中薄水铝石 a轴和 b轴的各向异性收缩、残留水或羟基离子的分布以及四面体位置空位的排列有关。事实上,一方面γ-A12O3的这些缺陷结构应该用四角点阵空间群中的一个,即中的最大子群来描述,它和晶格转移相一致;另一方面,类尖晶石结构的氧化铝的形成与氧化铝熔体的淬火或热氧化有关[43~45]。
3 介孔氧化铝的表面化学
研究氧化铝的表面化学,一方面有助于弄清氧化铝的表面酸性的实质,从而提高其作为载体所制备的催化剂的活性;另一方面有助于弄清氧化铝的表面结构,可揭示它与活性组分原子 (尤其是贵金属)间的密切关系。由于这与实际应用关系很大,因此被认为是今后工业研究的方向之一。
3.1 γ-A12O3和η-A12O3的表面结构
在氧化铝的众多晶型中,γ-A12O3和η-A12O3的晶体结构最为相似,两者均为尖晶石型结构,尖晶石的单位晶胞由 32个氧离子立方密堆积,在氧离子层间有 24个阳离子部位 (其中 16个为八面体,8个为四面体)统计地分布着 64/3个铝离子[46]。但γ-A12O3的前体多为拟薄水铝石,而η-A12O3的前体为三水氧化铝,前体的不同必然导致它们的表面结构有所差别。L evin等[47]认为,γ-A12O3和η-A12O3的结构中氧原子位于面心立方堆积中,并且在这两种晶型中存在缺陷尖晶石结构。他们认为在完美的尖晶石结构中,阳离子占据八面体结构的 1/2和四面体结构的 1/8。在γ-A12O3和η-A12O3中存在立方晶格的扭曲现象,并且有一些杂乱无序的阳离子空缺位,而γ-A12O3和η-A12O3结构的差别也就是晶格氧畸变程度的不同和阳离子间有序度的不同。选区电子衍射结果表明,由氢氧化物制得的η-A12O3是四角畸变的,轴比 c∶a(a,c为晶胞参数)在 0.985~0.993之间;而γ-A12O3畸变更严重,c∶a在 0.983~0.987之间[48]。另外,γ-A12O3的晶格氧比η-A12O3的晶格氧排列更为有序。这些差别与它们的前体不同相关。关于这种缺陷结构,Zhou等[49]用中子衍射谱对γ-A12O3和η-A12O3的精细结构进行了解析,他们认为两种晶相的表面层都存在异常的 32e氧配位的铝离子,但在η-A12O3的表面结构中不存在八重四配位的铝阳离子。该结果与γ-A12O3表面的分子动力学模拟结果一致[50]。
γ-A12O3的前体为拟薄水铝石,拟薄水铝石中的氧离子呈面心立方排列,在氢离子的参与下与临近层间的铝离子紧密堆积。而η-A12O3的前体三水氧化铝按化学成分可写成 A12O3·3H2O,即每个 A12O3分子结合 3个 H2O分子。但从结构上看 (见图 4),水并不是以 H2O分子的形式存在,而是以羟基基团的形式与铝离子结合。羟基呈六方最紧密堆积,铝离子填充于邻近的两层羟基离子之间的 2/3八面体空隙 (其余的空隙是空着的),组成配位八面体的结构层。每个铝离子与 6个羟基配位。这种紧密堆积的羟基构成一种层状结构,而相邻两层间以羟基所形成的氢键相连接[36]。

图 4 单层三水铝石结构[51]Fig.4 Structure of monolayer gibbsite[51].
3.2 介孔氧化铝的酸碱性及其表征
氧化铝表面的酸碱性对其催化活性影响很大,而氧化铝表面的酸碱性与其表面羟基密切相关。李慧等[52]分析了γ-A12O3催化剂表面羟基密度和酸强度对其催化性能的影响。对于氧化铝酸碱性的起因很多人进行了研究,也有人提出了一些设想和模型。如有人提出氧化铝表面的羟基是质子酸中心,而两个相邻的羟基脱水后构成 A l—O—A l桥键则为L酸中心。以前氧化铝表面的酸性主要是通过红外光谱法进行测定,人们认为氧化铝表面不存在B酸中心。但随着近代分析技术的发展,测定酸中心的方法越来越多,已有人用核磁共振谱证实了氧化铝表面有B酸中心存在。
Peri[53]还通过氨在γ-A12O3上的吸附来检测与催化活性相关的酸性位,用红外光谱法,通过氢 -氘交换、重量分析法和质谱分析法来辅助研究其表面吸附机制。此外还有一些实验表明,氧化铝不仅能使多核芳烃转变成相应的正离子自由基,还可以使四氰基乙烯 (电子接受体)转变成相应的负离子自由基。后一种现象表明,氧化铝表面上还存在着碱性中心,而且氧化铝失水过程中可能还不止形成一种碱性中心。至于氧化铝表面酸碱性的强弱,Peri[54]通过他提出的尖晶石型氧化铝表面结构的具体模型分析认为,脱水后孤立残存的氢氧根离子有 5种类型,它们以分别连接 0~4个氧离子而相区别。其中,连接 4个氧离子的氢氧根离子中心为最强的碱,而不直接与氧离子相连的氢氧根离子中心为最强的酸。根据氧化铝表面酸碱中心强弱不同会形成强弱不同的吸附中心[55]。酸性或碱性气体在温度较低时,能分别与较强和较弱的碱性或酸性中心形成吸附态;当升高温度,吸附分子接受热能大于吸附键能时,吸附分子脱离碱性或酸性表面而进入载气中,从而不能形成吸附态;只有较强的碱点或酸点才能与酸性或碱性气体形成吸附态。所以,不同温度产生的不同吸附态可表征固体表面酸碱强度。测定不同吸附态的量,可得到不同酸碱强度下酸碱中心的量,从而定量测量氧化铝表面的酸碱分布,并用热分析仪通过热量和质量的变化关联出γ-A12O3表面碱分布曲线和η-A12O3表面酸分布曲线。
3.3 介孔氧化铝表面铝的微环境
介孔氧化铝表面铝的微环境主要和与它相连的羟基和氧离子有关,因此与氧化铝所显示出来的L酸性和 B酸性相关。在 Peri[54]提出的尖晶石型氧化铝表面结构的模型中,就这个模型的 (100)晶面而言,对于干燥的氧化铝,表面的第一层由氧离子构成,其量只为第二层氧离子数的一半,第一层氧离子与第二层的铝离子相连接,有一半的铝离子裸露于表面上。第二层的氧离子数正好符合氧化铝的 A l与 O摩尔比。完全水合后的状态是:第二层的铝离子上都连接有羟基。脱水时,由两个相邻的羟基脱去一分子水,故脱水后尚残留一个羟基离子与一个裸露的铝离子。Peri[54]认为这个只与 3个氧离子相连的裸露的铝离子部位是活性中心,可以吸附水、氨、烃等多电子化合物。脱附时,质子转移到相邻的氧离子上形成羟基离子。所以铝离子因其缺电子特征而具有 L酸特征,而当其水合时则具有B酸特性。
另外,γ-A12O3催化剂中毒也与氧化铝表面铝的微环境有关。对于交换反应,活性中心可能是暴露的 A l3+相邻的羟基,它可因 CO2中毒而失活;而异构化中心不受 CO2影响[56]。
3.4 介孔氧化铝表面羟基结构
介孔氧化铝具有固体酸、碱的作用,而这些酸、碱特性是与其表面羟基结构分不开的。介孔氧化铝表面羟基还可因其制法、处理条件不同而分为“氢键键合羟基”和“孤立羟基”[57],氧化铝表面羟基的红外吸收波数 (见表 2)可用于区分不同的羟基。连业良等[58]的研究也支持了这种说法,他们认为即使是纯的氧化铝也会含有少量水,水随温度的变化可为羟基离子,也可为表面水分子,以氢键与下层表面相连或为多层物理吸附。可以通过红外光谱和重量分析法对介孔氧化铝的表面羟基结构进行研究。

表 2 不同氧化铝表面羟基的红外吸收波数[57]Table2 Infrared absorption wavenumber of hydroxyl groups on different alum ina surfaces[57]
Peri等[59]认为,即使经 1 000℃干燥后,γ-A12O3进一步加热时仍会有水析出,这些水和氧化铝的表面化学性质有关,并且会影响它的催化特性。H indin等[60]也认为γ-A12O3的催化活性和表面化学吸附水移去时形成的酸性位有关。Peri等[59]通过红外光谱分析发现,羟基基团和分子水都附着在氧化铝表面,加热到 400℃可移去所有的分子水,但会留下许多羟基基团。氧化铝在 650℃以上干燥,会在 3 698,3 737,3 795cm-1处产生 3个吸收峰,这些吸收峰与“孤立羟基”基团一一对应,3 698cm-1处的吸收峰所代表的羟基基团是这 3个羟基类型中酸性最强的。进一步观察发现这 3个羟基基团通过深度加热或氢 -氘交换会消失,但它们消失的速度不同。红外光谱的变化从扰动到消除或产生羟基带,这些羟基带强度的独立变化说明它们代表了在至少 3个不同类型的表面位的独特化学基团,这些基团在氧化铝催化反应中会发挥不同的作用。
Peri[61]后来通过红外光谱和重量分析法研究了γ-A12O3表面的水合作用,γ-A12O3的水合物表面每 1nm2含有 13个水分子,这些吸附水在高温加热时一部分会脱附,而另一部分会发生反应形成羟基基团。在 400℃以上移去羟基基团时会产生许多酸性位,γ-A12O3的许多重要催化特性都与这些酸性位有关。
4 结语
目前人们对于氧化铝的研究过多地集中于其微观或宏观层面,而对其介观层面的研究相对较少,有关氧化铝的表面性质还了解得不是很透彻。例如,对于γ-A12O3和η-A12O3等一些低温氧化铝的晶体结构仍不清楚;低温氧化铝的晶相定量分析仍很困难;氧化铝微晶形态与其晶型的关系尚不明确等。这些工作的开展必将有助于人们从氧化铝的微观结构入手,探讨其对宏观物性及其作为催化剂载体时对催化性能的影响。
另外,目前对有序介孔氧化铝的合成研究中还存在着各种问题,如合成工艺条件比较苛刻;合成的介孔尚未形成长程有序,并且孔结构的稳定性差;对有序介孔氧化铝的合成机理尚无合理解释等。如果能在有序三维连通孔道方面有所突破,将直接引导新一代活性氧化铝催化剂载体的产生,这将是氧化铝催化剂载体的一次重大革新。
无论从材料角度还是从催化应用前景来看,介孔氧化铝都是一种值得研究的介孔材料。相信随着这一领域的进一步研究,研究者会很快掌握合成介孔氧化铝材料的优良方法,真正发挥其独特优势,使其成为一种实用的优良催化材料。
1 James H A,Ender S.M orphological Control of Particles.Curr Opin Colloid Interface Sci,2000,5(1~2):160~167
2 Yang Q i,Deng Yida,Hu W enbin.Synthesis of A lum ina Nanofibers by a M ercury-M ediated M ethod.Ceram Int,2009,35(1):531~535
3 Yang Q i,Deng Yida,Hu W enbin.Preparation of A lum ina/Carbon Nanotubes Compositesby Chem ical Precipitation. Ceram Int,2009,35(3):1 305~1 310
4 张立岩,张鹏远,陈建峰.纳米纤维状γ-A l2O3粉体的制备与表征.石油化工,2004,33(3):240~243
5 Zhang Jun,Shi Fengjun,L in Jing,et al.Nanoparticles Assembly of Boehm ite Nanofibers W ithout a Surfactant.Mater Res Bull,2008,43(7):1 709~1 715
6 M ishra D,Anand S,Panda R K,et al.Effect of Anions During Hydrothermal Preparation of Boehm ites.MaterLett,2002,53(3):133~137
7 Guzman-Castillo M L,Hernandez-Beltran F,Fripiat J J,et al.Physicochem ical Properties of A lum inas Obtained from D ifferent A lum inum Salts.Catal Today,2005,107~108:874~878
8 Chen Xiangying,Lee S W.pH-Dependent Formation of Boehm ite(γ-A lOOH)Nnanorods and Nanoflakes.Chem Phys Lett,2007,438(4~6):279~284
9 Chen Xiangying,Zhang Zhongjie,L i Xueliang,et al.Controlled Hydrothermal Synthesis of Colloidal Boehm ite(γ-A lOOH)Nanorods and Nanoflakes and Their Conversion intoγ-A l2O3Nanocrystals.Solid State Commun,2008,145(7~8):368~373
10 Kaluza L,ZdrazilM,Zilkova N,et al.High Activity of Highly Loaded M oS2Hydrode Sulfurization CatalystsSupported on O rganized M esoporous A lum ina.Catal Commun,2002,3(4):151~157
11 陈增祥.汽车尾气催化剂介孔活性层载体材料的研究:〔学位论文〕.北京:北京工业大学,2005
12 Bagshaw S A,Pinnavaia T J.M esoporous A lum ina M olecular Sieves.Angew Chem,Int Ed,1996,35(10):1 102~1 105
13 Gonzalez-Pena V,D iaz I,M arquez-A lvarez C,et al.Thermally Stable M esoporous A lum ina Synthesized w ith Non-Ionic Surfactants in the Presence of Am ines.M icroporous M esoporous Mater,2001,44~45:203~210
14 D iaz I,Gonzalez-Peria V,M arquez-A lvarez C,et al.Transm ission ElectronM icroscopy Combined w ith Stochastic Reconstruction M ethods for Structural Characterization of Porous A lum ina Synthesized via Non-Ionic Surfactant-Templating Route.M icroporous M esoporous Mater,2004,68(1~3):11~19
15 Gonzalez-Pena V,M arquez-A lvarez C,Sastre E,et al.Synthesis of O rdered M esoporous and M icroporous A lum inas:Strategies for Tailoring Texture and A lum inum Coordination. Stud Surf Sci Catal,2002,142(2):1 283~1 290
16 Deng W,Bodart P,Pruski M,et al.Characterization of M esoporous A lum inaM olecularSieves Synthesized by Nonionic Templating.M icroporous M esoporous Mater,2002,52(3):169~177
17 Yang Peidong,Zhao Dongyuan,David I,et al.B lock Copolymer Templating Synthesis of M esoporous M etal Oxides w ith Large O rdering Lengths and Sem icrystalline Framework.Chem Mater,1999,11(10):2 813~2 826
18 Vaudry F,Khodabandeh S,DavisM E.Synthesis of Pure A lum ina M esoporous M aterials.Chem Mater,1996,8(7):1 451~1 464
19 郭建维,李龙焕,刘卅等.新型催化材料——介孔氧化铝分子筛的合成、表征以及应用前景.功能材料,2006,10(37):1 527~1 534
20 YadaM,OhyaM,M achidaM,et al.M esoporous Gallium Oxide Structurally Stabilized by Yttrium Oxide.Langmuir,2000,16(10):4 752~4 755
21 Spacil J. Production and App lication of Functionally G raded Cemented Carbide M aterials.Met Powder Rep,1998,53(2):41~42
22 YadaM,Ohya H,Ohe K,et al.Porous Yttrium A lum inum Oxide Templated by A lkyl Assemblies. Langmuir,2000,16(4):1 535~1 541
23 Valange S,DerouaultA,Barrault J,et al.One-Step Generation of Highly Selective Hydrogenation Catalysts Involving Sub-Nanometric Cu2O Supported on M esoporous A lum ina:Strategies to Control Their Size and D ispersion.J M ol CatalA:Chem,2005,228(1~2):255~266
24 李志平,赵瑞红,郭奋等.高比表面积有序介孔氧化铝的制备与表征.高等学校化学学报,2008,29(1):13~17
25 李志平,赵瑞红,郭奋等.复合模板剂制备有序介孔氧化铝.北京化工大学学报,2008,35(3):23~26
26 He Taobo,Xiang Lan,Zhu W ancheng,et al.H2SO4-Assisted Hydrothermal Preparation ofγ-A lOOH Nanorods.Mater Lett,2008,62(17~18):2 939~2 942
27 Gan Zhihong,N ing Guiling,L in Yuan,et al.M orphological Control of M esoporous A lum ina Nanostructures via Template-Free Solvothermal Synthesis.Mater Lett,2007,61(17):3 758~3 761
28 L iu Ye,M a D ing,Han Xiuwen,et al.Hydrothermal Synthesis of M icroscale Boehm ite and Gamma Nanoleaves A lum ina.M ater Lett,2008,62(8~9):1 297~1 301
29 M azloum i M,A ttarchi M,Lak A,et al.Boehm ite Nanopetals Self Assembled to Form Rosette-L ike Nanostructures.M ater Lett,2008,62(26):4 184~4 186
30 W u Xiuyong,W ang Debao,Hu Zhengshui,et al.Synthesis ofγ-A lOOH(γ-A l2O3)Self-Encapsulated and Hollow A rchitectures.M ater Chem Phys,2008,109(2~3):560~564
31 Guzman-Castillo M L,Bokhim i X,Toledo-Antonio A,et al.Effect of Boehm ite Crystallite Size and Steam ing on A lum ina Properties.J Phys Chem B,2001,105(11):2 099~2 106
32 赵琰.氧化铝(拟薄水铝石)的孔结构研究.工业催化,2002,10(1):55~63
33 L i W enjun,Shi Erwei,Zhong W eizhuo,et al.Grow th M echanism and Grow th Habit of Oxide Crystals.Cryst Growth,1999,203(1~2):186~196
34 Hong Tsai L in,L iu Hseh Tsang,Yeh Chuin Tih,et al.Electron M icroscopic Studies on Pore Structure of A lum ina.Appl Catal,A,1997,158(1):257~271
35 孔令斌.氧化铝及其水合物的结晶结构表征.石化技术与应用,2000,18(5):305~307
36 朱洪法编.催化剂载体制备及应用技术.北京:石油工业出版社,2002.348~349
37 Paglia G,Buckley C E,Rohl A L,et al.Boehm ite Derivedγ-A lum ina System.1.Structure Evolution w ith Temperature,w ith the Identification and Structural Determ ination of a New Transition Phase,y’-A lum ina.Chem Mater,2004,16:220~236
38 郝保红,黄俊华.晶体生长机理的研究综述.北京石油化工学院学报,2006,14(2):58~63
39 Tertian R,Papee D.Thermal and Hydrothermal Transformation of A lum ina.J Chem Phys,1958,55:341~353
40 Yanagida H,Yamaguchi G.Thermal Effects on the Lattices ofηandγ-A lum inum Oxide.Bull Chem Soc Jpn,1964,37:1 229~1 231
41 W ilson S J,M cConnell J D C.A Kinetic of the Systemγ-A lOOH/A l2O3.J Solid State Chem,1980,34(3):315~322
42 Tsuchida T,Furuichi R,Ishii T.Kinetics of the Dehydration of Boehm ites Prepared Under D ifferent Hydrothermal Conditions.Thermochim Acta,1980,39(2):103~115
43 Tayaram V,Levi C G.The Structure ofδ-A lum ina Evolved from the M elt and theγ→δTransformati.Acta M etal,1989,37(2):569~578
44 D ragoo A L,D iamond J J.Transitions in Vapor-Deposited A lum ina from300℃ to1 200℃.J Am Ceram Soc,1967,50(11):568~574
45 M orrisey K J,Czanderna K K,M errill R P,et al.Transition A lum ina Structures Studied Using HREM.U ltramicroscopy,1985,18(1~4):379~386
46 连业良,解韬青.催化用的氧化铝.青岛化工,1991,(2):20~23
47 Levin I,B randon D.M etastable A lum ina Polymorphs:Crystal Structures and Transition Sequences.J Am Ceram Soc,1998,81(8):1 995~2 012
48 L ippens B C,D e B oer J H.Study of Phase Transform ations During Calcinationo of A lum inum Hydroxides by Selected A rea Electron D iffraction.Acta Crystallogr,1964,17:1 312~1 321
49 Zhou R S,Snyder R L.Structures and Transformation M echanisms of theη,γ,andθTransition A lum inas.Acta Crystallogr,Sect B:Struct Sci,1991,47:617~630
50 B lonski S,Garofalini S H.M olecular Dynam ics Simulation ofα-A lum ina andγ-A lum ina Surfaces.Surf Sci,1993,295(1~2):263~274
51 Franks G V,Gan Yang.Charging Behavior at the A lum ina-W ater Interface and Implications for Ceram ic Processing.J Am Ceram Soc,2007,90(11):3 373~3 388
52 李慧,胡燚,苏国东等.合成方法对γ-A l2O3催化剂乙醇脱水性能的影响.石油化工,2009,38(4):373~378
53 Peri J B.Infrared Study of A dsorption of Amm onia on D ryγ-A lum ina.J Phys Chem,1965,69(1):231~239
54 Peri J B.A M odel for the Surface ofγ-A lum ina.J Phys Chem,1965,69(1):220~230
55 徐征,千载虎,崔圣范.测定固体催化剂表面酸碱分布新方法的研究.高等学校化学学报,1989,10(7):778~780
56 千载虎,李忆.复合氧化物酸性研究的进展.石油化工,1986,15(7):446~452
57 吴越编.催化化学 (下册).北京:科学出版社,1995.831~833
58 连业良,解韬青.水合氧化铝的制备.青岛化工,1992,(2):9~13
59 Peri J B,Hanna R B.Surface Hydroxyl Groups onγ-A lum ina.Phys Chem,1960,64(10):1 526~1 530
60 Hindin S G,W eller S W.Catalysis of Ethylene Hydrogenation and Hydrogen-Deuterium Exchangeby Dehydrated A lum ina. Adv Catal,1957,9:70~75
61 Peri J B.Infrared and Gravimetric Study of the Surface Hydration ofγ-A lim ina.Phys Chem,1965,69(1):211~219
Research Progress in Crystal Structure and Surface Chem istry of M esoporous Alum ina and Its Precursor
Guan Lili,Liu Yunqi,L i Guangci,L iu Chenguang
(State Key Laboratory of Heavy O il,Key Laboratory of Catalysis,CNPC,China University of Petroleum(East China),Q ingdao Shandong266555,China)
Research progresses in crystal structure and surface chem istry of m esoporous alum ina and its precursor w ere review ed. Starting from the m ain synthetic routes of m esoporous alum ina, its controlled synthesis w as system ically summ arized. In addition,new varieties of m esoporous alum ina w ith ordered pores different m orphologies and nano-crystalline alum ina assem blies w ere introduced in detail.The crystal grow th and control strategies,and the surface alum inum m icro-environm ent and the surface hydroxyl groups w ere discussed in detail.Hot issues and research opportunities in this field w ere prospected,and the suggestion of strengthening research in the alum ina m eso-levelw as proposed.
mesoporous alum ina;mesoporous alum ina precursor;crystal structure;surface chem istry
1000-8144(2010)07-0809-09
TQ133.1
A
2010-01-12;[修改稿日期 ]2010-05-06。
关丽丽 (1986—),女,河南省许昌市人,硕士生,电话0532-86984687,电邮 guanlixuchang@163.com。联系人:柳云骐,电话 0532-86981861,电邮 liuyq@upc.edu.cn。
国家重点基础研究发展计划项目 (2010CB226905);山东省自然科学基金项目(Y2008B58)。
(编辑 安 静 )
