多孔SiO2·xH2O负载RuB纳米粒子催化喹啉加氢反应
2010-11-06李贤均李瑞祥
张 磊 胡 博 陈 华 李贤均 李瑞祥
(四川大学化学学院,绿色化学与技术教育部重点实验室,成都 610064)
多孔SiO2·xH2O负载RuB纳米粒子催化喹啉加氢反应
张 磊 胡 博 陈 华 李贤均 李瑞祥*
(四川大学化学学院,绿色化学与技术教育部重点实验室,成都 610064)
通过水解,聚乙烯吡咯烷酮(PVP)保护,NaOH刻蚀等方法制备了多孔及富含表面羟基的SiO2·xH2O负载的RuB催化剂RuB/SiO2·xH2O,并用X射线衍射(XRD)、X射线光电子能谱(XPS)、透射电子显微镜(TEM)、傅里叶变换红外(FT-IR)光谱和BET(Brunauer-Emmett-Teller)等手段对该催化剂进行了表征.结果表明该催化剂具有良好的抗中毒能力,在3.0 MPa的H2压力和80℃的温和反应条件下,喹啉的转化率高于95%,生成1,2,3,4-四氢喹啉的选择性高于97%.并系统研究了表面羟基和溶剂对催化剂性能的影响,发现以水为溶剂时,RuB/SiO2· xH2O对喹啉加氢反应展示出较高的活性和对1,2,3,4-四氢喹啉较高的选择性,催化剂能够多次循环使用.这一体系的优异催化性能归属于载体表面羟基和水的协同作用.
钌;水合二氧化硅;喹啉;加氢;四氢喹啉
喹啉加氢可以生成1,2,3,4-四氢喹啉、5,6,7,8-四氢喹啉和十氢喹啉三种产物,反应如图1所示.
其中,1,2,3,4-四氢喹啉及其衍生物被广泛应用于药物中间体、农药、染料等的制备[1],催化加氢喹啉制备1,2,3,4-四氢喹啉是最简单有效的途径.喹啉加氢多以过渡金属络合物为催化剂,如[Rh(COD)Cl]2(COD:1,5-环辛二烯)[2]、[Rh2Cl2(COE)4/phosphine][3]、[Rh(COD)(PPh3)2]PF6[4]、[Ir(COD)Cl2][5-7]、[CpIrCl]2(Cp:环戊二烯基)[8]、[RuH(TPPTS)2(L)2](TPPTS:三磺化三苯基膦三钠盐)[9]、[RuH2(CO)(PBu)3]2[10]、Co(stearate)2-EtAl[11]等.这些催化剂在催化喹啉加氢反应中,反应条件温和,转化率和选择性都较高,但其价格昂贵,催化剂与产物的分离困难,难以实现工业应用.为此,研究催化剂与产物易于分离的多相催化剂体系对喹啉加氢显得尤为重要.人们研究过在水/有机两相体系中对喹啉加氢,但反应结果并不理想[12];Rh配合物与高聚物结合的均相催化剂多相化,则仍存在催化剂制备困难、金属流失难以解决的问题[13];直接将Ru负载于SiO2上催化喹啉加氢[14],生成1,2,3,4-四氢喹啉的选择性不高;用NiCl2-Li-naphthalene催化喹啉加氢,催化剂用量大,金属活性组分易中毒,催化剂不能循环使用[15];将纳米Ru负载于高分子材料上用于喹啉加氢,该催化剂抗中毒能力得到提高,但金属Ru的担载量高达10%(w,质量分数)[16].为解决含氮杂环化合物对催化剂的毒害作用,Vaccari等[17]通过在催化体系中加入质子酸来避免催化剂的中毒失活,然而质子酸的加入又会引起酸对设备的严重腐蚀.本课题组[18]曾报道水合载体ZrO2·xH2O负载的钌催化剂在水溶液中对催化喹啉加氢显示出较好的性能.但是干燥后的胶态催化剂Ru/ZrO2· xH2O中,水合二氧化锆比表面小,催化剂几乎没有孔道,活性金属中心大部分被包埋在载体内部,致使催化剂活性不佳,要获得高活性,Ru的用量仍然较大(6%(w)).为了既能提高催化剂活性,降低贵金属用量,又能利用载体表面羟基和溶剂水对喹啉加氢的促进作用,本文采用胶体保护和刻蚀以增加催化剂比表面积和孔径的办法,制备了一种多孔水合二氧化硅负载金属催化剂RuB/SiO2·xH2O,实现了低钌负载条件下对喹啉的高效加氢.
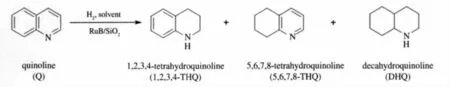
图1 喹啉加氢反应Fig.1 Hydrogenation of quinoline
1 实验部分
1.1 试 剂
喹啉(阿拉丁试剂公司)使用前经两次蒸馏后,加入少量盐酸,在室温下过滤,将滤液分馏,得含纯度99.0%的喹啉;RuCl3(昆明贵金属研究所)、原硅酸乙酯(TEOS)(Alfa Aesar,美国)、聚乙烯吡咯烷酮(PVP K15)(Fluka,瑞士)、聚乙二醇(PEG-200)(天津市瑞金特化学品有限公司)、氨水及其它试剂均为分析纯,使用前未经任何处理;氢气纯度大于99.99%.
1.2 催化剂的制备
将2.5 mL RuCl3水溶液(含20 mg的Ru)加入50 mL的二颈瓶中,同时加入15 mL的PEG-200,室温搅拌2 h,使Ru和PEG混合均匀,在搅拌下缓慢滴入5 mL含有60 mg NaBH4的水溶液,滴加完毕后继续搅拌30 min.然后旋转蒸发除去其中的水后,再加入3.7 mL的TEOS、20 mL异丙醇,继续搅拌1 h后滴入8 mL氨水,搅拌12 h.沉淀经离心分离,去离子水洗涤至中性,50℃真空干燥后得到催化剂前驱体RuB/SiO2·xH2O.
将上述催化剂前驱体置于25 mL二颈瓶中,加入4 mL去离子水和2 mL 5%PVP K15水溶液,搅拌回流1 h后,制得PVP保护的RuB/SiO2·xH2O.然后加入1 mL 5%NaOH水溶液对SiO2·xH2O刻蚀30 min,离心分离,固体物用去离子水洗涤至中性后, 50℃真空干燥得到含Ru 2%(w)的多孔RuB/SiO2· xH2O催化剂.其制备示意图如图2所示.
1.3 催化剂的表征
XRD在丹东方圆仪器有限公司DX-1000型X射线衍射仪上进行,使用Cu Kα射线,石墨单色器,管电压40 kV,管电流25 mA,扫描范围10°-80°,扫描速率0.03(°)·s-1,采样时间4 s.金属的分散度用美国FEI公司Tecnai G2 F20型电子显微镜观察,样品用乙醇分散.XPS在英国 KRATOS公司XSAM800型X光电能谱仪上进行,Mg KαX射线(hν=1253.6 eV);FT-IR在 Thermo Nicolet公司的NEXUS670FT-IR仪上分析.
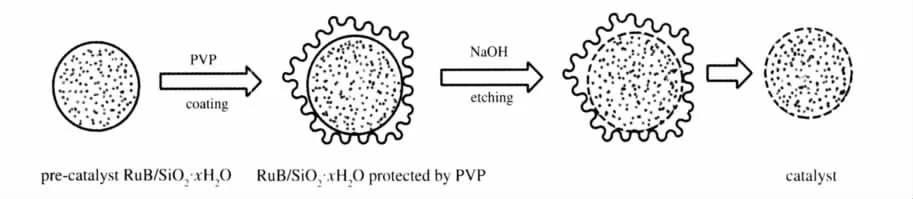
图2 催化剂制备示意图Fig.2 Process of catalyst preparation
1.4 催化剂的活性评价
加氢反应在带电磁搅拌的60 mL高压釜中进行,将所需要的溶剂、催化剂和底物分别加入反应釜中,然后用高纯氢气置换反应釜三次.再充氢气至所需的压力,升温至所设定的温度后,开始搅拌并记时.反应结束后,取出样品,离心分离催化剂.产物在GC920II型气相色谱仪(温岭市福立分析仪器有限公司)上分析,色谱柱为SE-30石英毛细管色谱柱(30 m×0.5 mm×0.25 μm,美国Supelco公司),氢火焰离子检测器,采用峰面积归一化法计算含量.
2 结果与讨论
2.1 催化剂的表征
催化剂的XRD谱图如图3所示,在2θ=43.67°处出现弱的金属钌的衍射峰,且衍射峰呈弥散状态,通过图4(a)的HRTEM图片可以看出,RuB纳米粒子在载体上分散的十分均匀,实验中使用的透射电子显微镜的加速电压是200 kV,电子的能量不足以穿透粒子较厚部分,因而使拍摄的图片左下角呈现一个黑色的色块.图4(b)是催化反应结束后的催化剂中纳米粒子的表征图片,我们选取了催化剂电子显微图像的局部进行了放大分析,图中圆形的轮廓为催化剂边缘,其中的小黑点为RuB纳米粒子,颜色较浅的部分为载体的显微图片;圆形轮廓外的视野为铜网背景.根据图4所示的催化剂的HRTEM照片可以估计出钌纳米粒子的粒径大约为2 nm.由谱图的选区电子衍射图像(SAED,selected area electron diffraction)可以看出,电子衍射图样为环状,证明催化剂中的纳米粒子以非晶态的RuB形式存在.
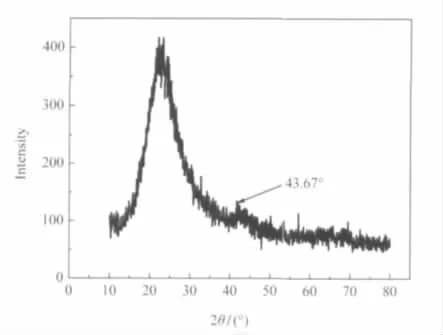
图3RuB/SiO2·xH2O催化剂的XRD谱Fig.3 XRD pattern of RuB/SiO2·xH2O catalyst
图5 为RuB/SiO2·xH2O催化剂的XPS谱,所有测试数据采用污染碳C 1s的结合能284.8 eV作为标准进行校正.Ru的3d5/2和3d3/2结合能分别为280.8和284.9 eV,比Ru0的相对应的理论值(280.0和284.1 eV)分别高0.8 eV,说明催化剂表面上的钌主要是介于Ru0和Ru+之间的低氧化态形式存在.

图4 RuB/SiO2·xH2O催化剂的HRTEM图Fig.4 High resolution transmission electron microscopy(HRTEM)images of RuB/SiO2·xH2O(a)image of catalyst before reaction,(b)image of catalyst after reaction;Insets are the corresponding selected area electron diffractions.NPs:nanoparticles
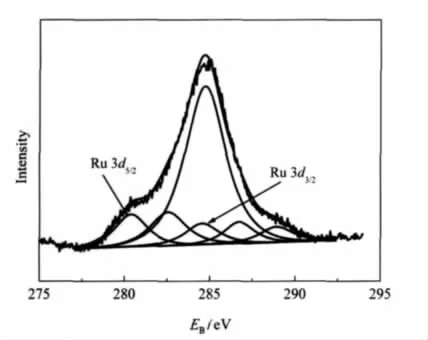
图5 RuB/SiO2·xH2O催化剂的XPS图Fig.5 XPS spectra of RuB/SiO2·xH2O
2.2 RuB/SiO2·xH2O对喹啉的催化性能
根据我们前面工作表明,Ru/ZrO2·xH2O表面是一种致密结构,许多金属活性位被包覆在载体内部,在催化反应时不能和底物作用,因此催化剂制备过程中需要高的金属负载量来提高催化活性.本文利用SiO2可以和碱反应的特性,将制备的胶态催化剂前驱体RuB/SiO2·xH2O用PVP保护,使用NaOH水溶液为刻蚀剂来刻蚀SiO2,由于催化剂被PVP包覆的部分被保护,没有包覆的部分NaOH将和SiO2反应产生一些孔道,使包埋在水合二氧化硅内部的活性组分暴露到催化剂表面.这样制备的催化剂不需要经焙烧使其脱水产生孔道,既保证了催化剂表面羟基的存在,又增加了催化剂活性中心在催化剂表面的数目,以实现低金属负载量催化剂的高催化活性.
2.2.1 溶剂对加氢反应的影响
液相反应中,溶剂起着十分重要的作用,不同的溶剂,由于和催化剂的吸附性能不同,以及与反应物或产物的作用不同,将导致对催化反应的转化率和选择性产生极大的影响.表1列出了几种常见的溶剂对多孔RuB/SiO2·xH2O催化喹啉加氢转化率和选择性的影响
表1数据表明,催化剂在水中显示出最好的活性和选择性,喹啉的加氢活性顺序为水>醇>醚,这与López-Linares[2]和Sánchez-Delgado[16]所使用的催化剂在不同溶剂中对喹啉加氢活性报道的结果一致.相同的温度、压力下,以水为溶剂,反应仅3 h,喹啉的转化率就能达到97.3%,比在EtOH中的加氢速度要高得多,这和水合ZrO2负载的钌催化剂表现出相似的性质[18],根据文献报道[19],ZrO2表面存在大量的羟基.本课题组对喹啉加氢研究中提出了载体表面羟基和溶剂水对喹啉加氢可能的协同作用机理,但没有得到有关底物和表面羟基作用的证据[18].由于通过TEOS水解生成的SiO2·xH2O胶体表面含有丰富的表面羟基,以胶态的SiO2·xH2O作为催化剂载体制备催化剂可以进一步证明我们的推论.图6为RuB/SiO2·xH2O催化剂的IR谱.从图6可以看出,催化剂表面吸附喹啉后,表面羟基的伸缩振动频率由3436 cm-1减少到3434 cm-1,羟基的弯曲振动吸收从1632 cm-1降低到1621 cm-1,说明催化剂表面的羟基和喹啉之间形成了氢键.氢键的形成促进了底物在催化剂表面的吸附,同时溶剂水又能和碱性更强的加氢产物形成更强的氢键,与此同时,喹啉加氢生成的1,2,3,4-四氢喹啉在水溶液中的溶解度比喹啉大[20],这两个因素同时促进了加氢产物在催化剂表面的脱附,从而提高催化剂活性和抗中毒能力.同样,溶剂对反应的选择性也有较大的影响,加氢生成1,2,3,4-四氢喹啉的选择性遵循质子溶剂大于非质子溶剂的原则.非质子溶剂中,喹啉加氢生成5,6,7,8-四氢喹啉和十氢喹啉的选择性要明显高于质子溶剂,如在甲苯和四氢呋喃分别作为喹啉加氢的反应溶剂时,生成5,6,7,8-四氢喹啉和十氢喹啉的选择性之和分别为11.1%和11.9%;这一结果也是氢键作用的一个有力证据,同时Fish等[21-25]对喹啉均相加氢机理的研究也可以作为氢键作用的另一个有力证据.该催化剂体系中喹啉和催化剂在不同溶剂中的氢键作用如图7所示.溶剂为非质子溶剂时,载体表面的羟基除了可以和N原子形成氢键外,也能和喹啉分子中苯环上的大π键电子形成氢键[14],从而导致活化氢分子对该环进攻,生成5,6,7,8-四氢喹啉;水做溶剂时,水和苯环的氢键作用抑制了喹啉分子中的苯环和表面羟基的相互作用,使喹啉分子中的苯环难以吸附到催化剂表面以及受到催化剂表面活化氢分子的进攻,致使生成5,6,7,8-四氢喹啉变得困难,导致生成1,2,3,4-四氢喹啉的选择性大幅度提高.

表1 溶剂对喹啉加氢反应的影响Table 1 Influence of solvents on hydrogenation of quinoline

图6 RuB/SiO2·xH2O催化剂的IR谱Fig.6 IR spectra of RuB/SiO2·xH2O catalyst
2.2.2 催化剂焙烧温度对其结构与催化性能的影响
为了进一步考察催化剂表面性质对催化性能的影响,我们研究了催化剂焙烧温度变化对其结构和性能的影响.将制备的多孔RuB/SiO2·xH2O在不同的温度下焙烧1 h后,对催化剂进行了比表面积、孔体积、孔径以及催化性能测试,结果如表2所示.
从表2的数据可以看出,通过NaOH刻蚀、未经高温焙烧的催化剂RuB/SiO2·xH2O具有大的比表面积、孔体积和小的孔径.随着焙烧温度的升高,催化剂的比表面积和孔体积逐渐减小,孔径逐渐增加,催化活性随焙烧温度升高而下降.焙烧温度升高,载体脱水使表面羟基损失;这样,不仅使催化剂表面羟基和水对催化反应的协同作用被削弱甚至消失,而且破坏了催化剂的多孔特性,使小孔在焙烧过程中融合成大孔,导致比表面积下降,催化活性降低.同时,焙烧温度增加,金属钌纳米粒子会团聚长大,降低催化剂的活性.作为对比,我们用商品SiO2作为载体制备了相同负载量的RuB/SiO2对喹啉进行催化加氢,实验结果表明,相同反应条件下,商品SiO2做为载体制得的催化剂加氢喹啉生成1,2,3,4-四氢喹啉的转化率仅为58.3%,这是因为普通的SiO2经焙烧,表面缺少羟基不能和水形成协同作用.
2.2.3 温度、压力对加氢的影响
以H2O为溶剂,在3.0 MPa氢气压力,反应3 h的条件下,考察了反应温度对RuB/SiO2·xH2O催化喹啉加氢反应的影响,结果如表3所示.温度低于70℃,喹啉的转化率随着温度的升高而快速增加,此时喹啉加氢生成1,2,3,4-四氢喹啉的选择性变化不明显,维持在98%左右,当温度升到80℃时,转化率已经达到了97.3%.文献报道[12-13],在3 MPa的压力下,温度甚至达到140℃时,喹啉的转化率才能达95%,可见本文的反应条件更加温和.

图7 RuB/SiO2·xH2O催化剂在不同溶剂中和喹啉的氢键作用Fig.7 Hydrogen bonding between RuB/SiO2·xH2O catalyst and quinoline in different solvents(a)protic solvent,(b)aprotic solvent

表2 催化剂的灼烧温度对喹啉加氢反应的影响Table 2 Effect of catalyst calcination temperature on hydrogenation of quinoline

表3 反应温度对喹啉加氢的影响Table 3 Effect of reaction temperature on hydrogenation of quinoline
表4显示了氢气压力对喹啉加氢反应的影响.可以看出,该催化体系对氢气压力变化并不敏感,在80℃,即使在0.5 MPa的低压条件下,喹啉加氢的转化率也能达到90.0%.该结果也比许多文献报道结果好许多,如文献报道[16],在10%Ru/P4Vpy(聚乙烯基吡啶))催化喹啉反应中,在120℃,H2压力4 MPa的条件下,反应转化率仅为50%.
2.2.4 底物/钌的摩尔比对加氢反应的影响
为了尽可能减少催化剂的使用量,我们考察了底物与钌的摩尔比变化对喹啉加氢反应的影响,结果列于表5中.可以看出,催化剂的用量对喹啉加氢的活性和选择性影响较小.在考察的范围内,催化剂对1,2,3,4-四氢喹啉保持了高的选择性,TOF(转化频率)值随着底物/钌摩尔比的增加由64.7 h-1增加到115.5 h-1,TON(转化数)值由194.6增加到346.4,当底物/钌摩尔比增加到800∶1,反应时间为3 h时, TON值仍为344.8.将反应时间延长到8 h后,TON值由344.8增加到了733.6.本课题组制备的非多孔催化剂6%Ru/ZrO2·xH2O[18],当底物/钌摩尔比由250∶1增加到500∶1时,TON值由240仅增加到314,进一步增加到1000∶1时,TON值反而下降到了288.而本文制备的催化剂在考察范围内的TON值都保持了一个稳定的结果,说明了本文制备的催化剂十分稳定,表现出良好的抗中毒能力.同时说明催化剂的用量有一个合适的范围,在选择最佳的反应条件下,本文制备的催化剂可以在保持高催化活性和高选择性的同时获得较快的反应速率.

表4 氢气压力对喹啉加氢反应的影响Table 4 Effect of hydrogen pressure on hydrogenation of quinoline
2.2.5 催化剂的循环使用

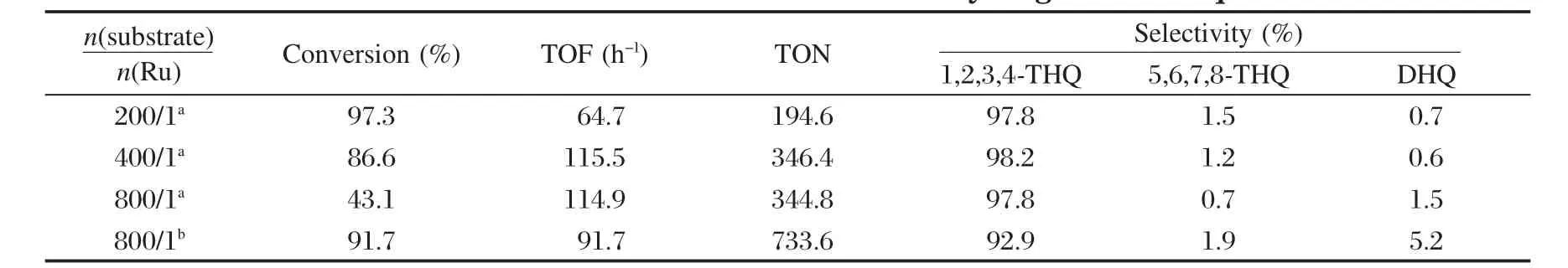
表5 底物/钌的摩尔比对喹啉加氢反应的影响Table 5 Influence of substrate/Ru molar ratio on hydrogenation of quinolone
RuB/SiO2·xH2O催化剂催化喹啉加氢反应中循环总结如图8所示,从图8可以看出,RuB/SiO2· xH2O催化剂在第三次使用中转化率有所下降,但是仍然取得88.3%的转化率,在第四次循环中催化剂活性发生了明显降低,转化率下降到了62.4%.有报道[21]认为这是由于催化剂在使用过程中金属纳米粒子团聚或者喹啉及其加氢中间体和金属活性物种配位作用加强而使催化剂的活性降低,La Vopa等[26]报道喹啉加氢过程中氮原子与催化剂的吸附能力大小顺序:喹啉<1,2,3,4-四氢喹啉<5,6,7,8-四氢喹啉<十氢喹啉,如果这些含氮化合物不能从催化剂上脱附,将引起催化剂中毒失活,从图4(b)中可以看出,催化剂中的金属粒径在反应前后没有明显的变化,说明本文中催化活性下降的原因不是由于金属纳米粒子团聚导致的,同时用诱导耦合等离子发射光谱(ICP)测定反应后水溶液中Ru的含量表明金属Ru的流失率为0.08%,证明了催化活性下降是由于反应中使用的催化剂量过少,在催化剂和反应底物分离循环过程中不可避免地由于催化剂的流失而造成了催化剂活性的下降.以上结果进一步表明本文制备的催化剂有较好的抗氮中毒的能力.
3 结论
通过水解刻蚀制备了低金属负载量,且富含表面羟基的多孔催化剂RuB/SiO2·xH2O.以水为溶剂,在3.0 MPa和80℃条件下,该催化剂对喹啉加氢转化率高于95%,其中1,2,3,4-四氢喹啉的选择性大于97%.该催化剂对喹啉加氢不仅显示出优异的催化活性,而且抗中毒能力强.
1 Katritzky,A.R.;Rachwal,S.;Rachwal,B.Tetrahedron,1996,52: 15031
2 Alvarado,Y.;Busolo,M.A.;López-Linares,F.J.Mol.Catal.AChem.,1999,142:163
3 Rosales,M.;Vallejo,R.;Soto,J.J.;Chacón,G.;Gonález,Á.; González,B.Catalysis Letters,2006,106:3
4 Sánchez-Delgado,R.A.;Rondón,D.;Andriollo,A.;Herrera,V.; Martín,G.;Chaudret,B.Organometallics,1993,12:4291
5 Wang,W.B.;Lu,S.M.;Yang,P.Y.;Han,X.W.;Zhou,Y.G. J.Am.Chem.Soc.,2003,125:10536
6 Yang,P.Y.;Zhou,Y.G.Tetrahedron:Asymmetry,2004,15:1145
7 Lu,S.M.;Han,X.W.;Zhou,Y.G.Adv.Synth.Catal.,2004,346: 909
8 Fujita,K.;Kitatsuji,C.;Furukawa,S.;Yamaguchi,R.Tetrahedron Letters,2004,45:3215
9 Rosales,M.;Castillo,J.;González,A.;González,L.;Molina,K.; Navarro,J.;Pacheco,I.;Pérez,H.Trans.Metal Chem.,2004,29: 221
10 Busolo,M.A.;López-Linares,F.;Andriollo,A.;Páez,D.E. J.Mol.Catal.A-Chem.,2002,189:211
11 Frediani,P.;Pistolesi,V.;Frediani,M.;Rosi,L.Inorganica Chimica Acta,2006,359:917
12 Rojas,I.;López-Linares,F.;Valencia,N.;Bianchini,C.J.Mol. Catal.A-Chem.,1999,144:1
13 Bianchini,C.;Frediani,M.;Mantovani,G.;Vizza,F. Organometallics,2001,20:2660
14 Bianchini,C.;Dal Santo,V.;Meli,A.;Moneti,S.;Moreno,M.; Oberhauser,W.;Psaro,R.;Sordelli,L.;Vizza,F.J.Catal.,2003, 213:47
15 Alonso,F.;Yus,M.Adv.Synth.Catal.,2001,343:188
16 Sánchez-Delgado,R.A.;Machalaba,N.;Ng-a-qui,N.Catal. Commun.,2007,8:2115
17 Campanati,M.;Cassgrande,M.;Fagiolino,I.;Lenarda,M.; Storaro,L.;Battagliarin,M.;Vaccari,A.J.Mol.Catal.A-Chem., 2002,184:267
18 Zhang,R.M.;Fan,G.Y.;Li,C.;Wang,Y.Y.;Li,R.X.;Chen,H.; Li,X.J.Acta Phys.-Chim.Sin.,2008,24:965 [张瑞敏,樊光银,李 诚,王瑛瑛,李瑞祥,陈 华,李贤均.物理化学学报, 2008,24:965]
19 Ma,Z.Y.;Xu,R.;Yang,C.;Wei,W.;Li,W.H.;Sun,Y.H.Acta Phys.-Chim.Sin.,2004,20:1221 [马中义,徐 润,杨 成,魏 伟,李文怀,孙予罕.物理化学学报,2004,20:1221]
20 Lide,D.R.Handbook of chemistry and physics.Boca Raton,FL: CRC Press/Taylor and Francis Group,version 2010
21 Fish,R.H.;Kim,H.S.;Fong,R.H.Organometallics,1989,8: 1375
22 Fish,R.H.;Michaels,J.N.;Moore,R.S.;Heinemann,H.J.Catal., 1990,123:74
23 Fish,R.H.;Baralt,E.;Kim,H.S.Organometallics,1991,10: 1965
24 Fish,R.H.;Kim,H.S.;Fong,R.H.Organometallics,1991,10: 770
25 Baralt,E.;Smith,S.J.;Hurwitz,J.;Horváth,I.T.;Fish,R.H. J.Am.Chem.Soc.,1992,114:5187
26 La Vopa,V.;Satterfield,C.N.J.Catal.,1988,110:375
Catalytic Performance of Porous SiO2·xH2O Supported RuB Nanoparticles for the Hydrogenation of Quinoline
ZHANG Lei HU Bo CHEN Hua LI Xian-Jun LI Rui-Xiang*
(Key Laboratory of Green Chemistry and Technology of the Ministry of Education,College of Chemistry,Sichuan University, Chengdu 610064,P.R.China)
A porous and hydroxyl group-rich catalyst RuB/SiO2·xH2O was prepared by hydrolyzing ethyl silicate, protecting SiO2·xH2O with polyvinyl pyrrolidone(PVP),and etching SiO2·xH2O with NaOH.The catalyst was characterized by X-ray diffraction(XRD),X-ray photoelectron spectroscopy(XPS),transmission electron microscopy (TEM),Fourier transform infrared(FT-IR)spectroscopy,and BET(Brunauer-Emmett-Teller).We found that the catalyst showed excellent performance for the hydrogenation of quinoline under mild condition.At a hydrogen pressure of 3.0 MPa and a reaction temperature of 80℃,the conversion of quinoline reached 95%and the selectivity for 1,2,3,4-tetrahydroquinoline was 97%.This porous catalyst also showed an excellent anti-poisoning characteristic.The catalyst can be reused several times.We also investigated the effect of surface hydroxyl groups and the solvent on catalytic activity and selectivity.The results showed that using water as a solvent leads to higher catalyst activity and selectivity for the hydrogenation of quinoline.The mechanism of quinoline hydrogenation over the catalyst is discussed.The coordination of the nitrogen on quinoline onto the surface of ruthenium nanoparticles,the effect of hydrogen bond among the surface hydroxyl groups of the catalyst and the nitrogen present in quinoline and in the water solvent were favorable for the adsorption of the substrate and the desorption of the products from the surface of the catalyst.
Ruthenium;SiO2·xH2O;Quinoline;Hydrogenation;Tetrahydroquinoline
O643
Received:February 19,2010;Revised:May 24,2010;Published on Web:July 6,2010.
*Corresponding author.Email:sculiruixiang@163.com;Tel/Fax:+86-28-85412904.
The project was supported by the National Natural Science Foundation of China(21072138).
国家自然科学基金(21072138)资助项目
ⒸEditorial office of Acta Physico-Chimica Sinica
