阳离子抗菌肽分子设计的研究现状
2010-11-02王联结
王 威,王联结
(陕西科技大学生命科学与工程学院,陕西西安 710021)
阳离子抗菌肽分子设计的研究现状
王 威,王联结*
(陕西科技大学生命科学与工程学院,陕西西安 710021)
随着传统抗生素的大量使用,许多病原微生物对抗生素产生了耐药性。不断寻找新一代的抗菌药物已经成为制药科研工作者的共有目标。阳离子抗菌肽具有相对分子量小、抗菌谱广、热稳定性好和抗菌机理独特等优点,有望成为新一代的抗菌药物。最大限度地提高其活性和稳定性、降低毒性和成本,是抗菌肽新药开发的首要问题。用分子设计手段改造抗菌肽已成为解决这一问题的关键。国内外的研究者在抗菌肽的分子设计方面做出了大量的努力,本文对抗菌肽的人工设计方法进行综述,分析了各种设计方法的优缺点,同时对抗菌肽分子设计的发展前景进行了展望。
抗菌肽,两亲α-螺旋,分子设计
1 抗菌肽人工设计的原则
1.1 阳离子性和两亲性原则
阳离子抗菌肽在序列和结构上表现出多样性,但其分子设计都有两个共同的要求,即阳离子性和两亲性[3]。前者决定其能否选择性地与显负电性的细菌细胞膜外表面相互吸引,而不与呈中性的真核生物细胞膜外表面产生相互作用,这也就是它们对哺乳动物细胞没有毒害作用的主要原因[4];后者决定其能否有效插入细菌细胞膜内,形成疏水通道。但是这也有例外,例如:来源于成熟雄鱼精巢精细胞的鱼精蛋白,由于序列中精氨酸含量在 60%以上,其二级结构并不呈现两亲α-螺旋和两亲β-折叠结构,因此在鱼精蛋白序列的改造过程中应更多考虑其抗菌肽机理。实际上,阳离子抗菌肽并不要求严格的一级和二级结构,不同序列类型也可形成具有两亲性的结构而与细胞膜结合[5]。
1.2 结构简化原则
在保证抗菌活性前提下,应尽量简化阳离子抗菌肽的序列和结构,一方面易于进行合成降低成本,另外一方面也有助于阐明单一因素改变对活性的影响。例如,序列当中避免出现 Cys,以防合成序列形成二硫键而影响抗菌肽的两亲α-螺旋结构[6]。
2 阳离子抗菌肽的设计方法
阳离子抗菌肽分子设计是指基于结构与功能关系,通过一系列生物信息学方法,实现对天然阳离子抗菌肽的定向改造或全新设计,获得更加符合需要的非天然序列,并阐明其活性机制,供进一步开发利用。在阳离子抗菌肽设计的过程中,生物信息学起到了极其重要的作用。众多的生物软件和网络在线工具给科研工作者提供了极大的方便。利用 NCB I的 ClustalX程序和网络在线工具 http:// bioinfo.genotoul.fr/multalin/multalin.h tml,以 Fasta格式输入多个抗菌肽分子序列,序列比对得到抗菌肽分子的一级保守序列;利用生物软件DNAstar、Vector NTI Advance、ANTHEPROT和网络工具 http:// alexander.compbio.ucsf.edu/~nomi/nnpredict.h tml进行序列的二级结构分析;利用 ANTHEPROT软件的Hydrophobicity计算出该肽段亲水和疏水位点,用HelicalWheel获得三维螺旋轮结构,并分析螺旋两侧和整体的亲水和疏水状态;Ras Mol和 Vector NTI Advance等软件能够绘出多肽的三维结构供研究者参考。
到目前为止,国内外学者使用不同的设计方法对阳离子抗菌肽进行序列的改造,总结起来可以分为四种:基于抗菌肽结构与功能的设计方法、基于抗菌肽抗菌机理的设计方法、抗菌肽氨基酸残基侧链的化学修饰和其他设计方法。
2.1 基于抗菌肽结构与功能的设计方法
抗菌肽种类繁多,结构上以三种类型为主:形成α-螺旋的抗菌肽;含 Cys形成二硫键的抗菌肽;富含1~2种氨基酸的抗菌肽,例如富含 Pro和 His[7]。抗菌肽的结构功能研究以及分子设计,目前大部分集中于两亲α-螺旋抗菌肽,原因在于α-螺旋抗菌肽分布最广,数量最多,并具有广谱抗性;该类抗菌肽长度短(<40个残基),易于化学合成;线性结构简单,易于通过圆二色谱分析和多维核磁共振进行结构测定[6]。基于以上特点,两亲α-螺旋抗菌肽已成为分子设计的首选对象。因此,在众多的抗菌肽分子设计方法中,以天然序列为模板,维持抗菌肽两亲α-螺旋结构不变,取得了很大的成就。
2.1.1 残基替换 残基替换就是替换天然序列中的一个或多个氨基酸残基。Song Yub Shin[8]等人将天然的α-螺旋抗菌肽 K6L6W(K WKKLLKKLLKLLNH2)的Leu6替换成 Pro(K WKKLPKKLLKLL-NH2),残基替换后的 K6L6W表现出的抗菌活性可以和蜂毒肽相媲美,然而其溶血活性远低于蜂毒肽。同时证明了 Pro出现在两亲α-螺旋抗菌肽中,对抗菌肽的选择性毒性起到了完美的效果。Sawai[9]等在保留两亲α-螺旋结构的前提下进行单残基突变,用 Gly取代抗菌肽 Ovispirin-1(KNLRR II RKII H IIKKYG)第 10位的 Ile,结果克服了对红细胞的溶血活性,并提高了抗菌活性。Yanga[10]等研究了富含 Arg和 Pro的抗菌肽 Tritrpticin,他们将抗菌肽序列中 Arg替换为 Lys,发现虽然两者的构象没有显著的差别,但替换后的抗菌肽活性提高了 2倍而且溶血活性大大降低。
国内的葛晓东[11]等人将 LL-37的 Glu16、Asp27和Glu36用 Gln、Asn、Gln所替换,在不改变 LL-37两亲α-螺旋结构的前提下,增加了序列的净电荷,实验证明,替换后序列的抗菌活性有所增加,同时降低了LL-37的溶血活性。
2.1.2 截取天然抗菌肽的部分序列 众所周知,人源性的LL-37及 GKE两个抗菌肽就是很好的例子。人类唯一的 Cathelicidin Gene所编码的蛋白质为hCAP18(human cationic ant imicrobial protein 18),其中 hCAP18104-140为长度 37个氨基酸的多肽 LL-37[11],具有接近完美的两亲α-螺旋结构,具有抗菌活性同时具有溶血活性。Thorgerdur Sigurdardottir[12]等人截取LL-37的不同部分,分别命名为LLG、GKE、FKR,这三个序列都具有正电性和两亲α-螺旋结构。实验结果表明,LL-3714-34的 GKE有着与 LL-37相同甚至更好的抗菌活性,同时 GKE的溶血活性较LL-37低。
2.1.3 序列模板法 Tossi[13]等人收集了 85条来自于昆虫、青蛙和哺乳动物的能形成两亲α-螺旋抗菌肽。通过对N-末端的 20个氨基酸残基进行序列比对和统计每个位点氨基酸残基出现的概率,得出序列模板:Ghhp+hxpxh+phh+xhhxh(G:Gly,h:疏水性氨基酸,+:碱性氨基酸,p:极性的或带电荷的氨基酸,x:不确定的氨基酸)。Tossi等设计出带 7个正电荷的抗菌肽 PGAa(G ILSKLFKALKKAAKHAAKANH2)。PhD程序预测该序列能形成两亲α-螺旋结构。抑菌实验表明,PGAa不仅具有对 G+和 G-细菌的抑菌活性,而且表现出对实验中的真菌具有抑菌活性。Tiozzo[14]等以 Tossi等人收集的能形成两亲-α螺旋的抗菌肽的序列模板为基础,设计出类似于哺乳动物中抗菌肽 Cathelicidin家族的新型肽 PGYa (GLLRRLRDFLKKIGEK FKKIGY-NH2)。同时,参考昆虫和两栖动物中抗菌肽的特征,设计出类似物PGAa(G ILSKLGKALKKAAKHAAKA-NH2)。通过抑菌实验,发现 PGYa对 G-和 G+细菌均有抗性,而PGAa则表现对真菌假丝酵母有抗性。
2.1.4 组合化学库方法 Sung Yu Hong[15]等人通过统计分析,采用在两亲α-螺旋结构中出现频率较大,对结构贡献重要的 7种氨基酸 (Lys、Leu、Val、Phe、Ser、Gln和 Pro)作为原料,化学合成了各种组合的十肽类似物,并从中筛选出 K LS2(KK VVFKFKFK-NH2)。K LS2对细菌具有广谱抗菌性,且没有溶血性。
2.1.5 螺旋轮方法 螺旋轮方法就是设计疏水性和亲水性氨基酸残基交替出现在一条序列里,以期形成两亲α-螺旋结构。Fernandez[16]等利用螺旋轮方法,得到仅有8个残基的α螺旋肽链(K QQRWLWLW)具有强抑菌作用。
2.2 基于抗菌肽抗菌机理的设计方法
Yechiel Shai[17]和 Ziv Oren[17]基于地毯式抗菌机理和抗菌肽形成低聚物的特性,设计了一系列的新型抗菌肽(分子中既有 D-氨基酸,也有L-氨基酸),这种方法为未来抗菌肽的分子设计提供了一个新的突破口。南京工业大学的陈菲[18]等人根据颗粒溶素(granulysin)的抗菌机理,维持天然序列α-螺旋结构不变的情况下,Asp43替换为 Thr以减小片段的负电荷;Asn47替换为Arg以增加片段的正电荷和亲水性; Tyr52替换为 Phe以增加片段 C末端的疏水性。实验证明,新肽在抗菌机理方面能更好的通过静电作用扰乱 G+细菌的细胞壁和细胞膜结构。

表 1 抗菌肽设计方法的优点与缺点比较
2.3 抗菌肽氨基酸残基侧链的化学修饰
2005年,Potter[19]等在研究鱼精蛋白与细菌间静电荷的相互作用时,用 1,2-cyclohexanedione部分修饰精氨酸的胍基,降低了鱼精蛋白的正电荷。当被修饰的精氨酸残基数目少于 4个时,鱼精蛋白对细菌的最低抑菌浓度 (M I C)值有所降低。Sangita Roy[20]等人化学合成了一个带正电荷的二肽类似物,该两亲性物质通过形成空间网状结构的凝胶而起到抗菌作用,研究者通过改变侧链烷基的长度选择抗菌活性最高的凝胶。日本东京大学[21](Tokyo university)的学者通过在鱼精蛋白的一端添加非氨基酸组分,希望增加鱼精蛋白的乳化能力。
2.4 其他设计方法
除了上述的设计方法以外,还有多种设计方法。例如:
a.D-型氨基酸替代相应L-型氨基酸:D-型氨基酸的化学性质和L-型氨基酸一样,这样就不会改变抗菌肽的阳离子性和两亲性,但是 D-型氨基酸的存在降低了蛋白酶对抗菌肽的水解作用。
b.使用约束氨基酸,这样能够克服多肽的固有柔性,同时把约束氨基酸引入生物活性肽的氨基酸序列,形成的局部约束以及约束由共价或非共价立体相互作用所决定的侧链构像[22]。
c.将两条抗菌肽连接起来,希望集二者优点于一身的杂合肽设计方法。例如抗菌肽 CecropinA抗细菌能力强,而抗菌肽Magainin抗真菌能力较强,能否各取所长并克服原有毒性呢?为实现这个设想, Shin[23]等将 CecropinA的 N端 8个残基作头, Magainin的N端 12个残基作尾,合成得到了杂合链CecropinA(1~8)-magainin(1~12)(缩写 CE-MA),发现其表现出很强的抗细菌活性,且没有溶血活性。
3 各种抗菌肽设计方法比较
对于不同来源和不同序列特征的抗菌肽,采取了不同的方法对其进行设计和改造,这些方法有着不同的优缺点,各种设计方法的比较见表 1。
4 总结及展望
在阳离子抗菌肽的分子设计中,如何提高抗菌活性和选择性毒性是最为关键的问题。结构与功能关系的研究表明,影响抗菌肽抗菌活性和选择性毒性的因素主要有 5个,即构象(χ)、电荷 (Q)、两亲性(A)、疏水性 (H)和亲疏水面的夹角 (θ)。这 5个因素之间相互依赖,改变其中一个因素就会对其他因素产生影响[24]。图 1表示了它们之间的相互关系。在这 5个因素当中,电荷和疏水性对抗菌肽的活性和选择性毒性作用最为明显。
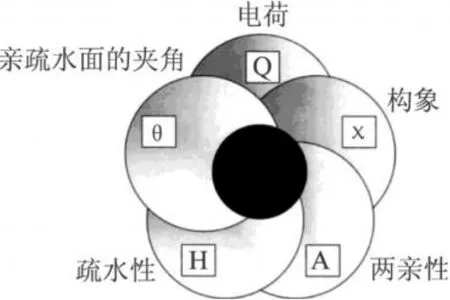
图 1 5个因素之间的相互关系
4.1 电荷
阳离子抗菌肽的电荷可直接影响其活性和选择性毒性。可通过改变肽链长度、取代肽链中 Arg和Lys、C末端酰胺化和增加酸性氨基酸等方法合成模拟肽,并研究电荷对其活性的影响。Margitta Dathe[25]等人通过改变正电荷筛选抗菌活性和选择性毒性最优的 magainin抗菌肽的实验证明,当阳离子抗菌肽的电荷小于 +5时,抗菌活性随正电荷增加而增加;当电荷大于 +7时,增加正电荷对活性影响不大,但抗菌肽的溶血活性增加并且选择性毒性降低。Helena Hujakka[26]的实验也证明了电荷对抗菌肽活性的影响至关重要。实验中的三种抗菌肽的比较见表2。
抑菌实验表明,在 5~10μmol/L的浓度下,TA和TAd完全抑制金黄色葡萄球菌的生长,然而 T Ac没有表现出任何的抗菌活性。10μmol/L的浓度下,Tad能够完全抑制大肠杆菌的生长,就算浓度增加到20μmol/L,TA也没表现出其对大肠杆菌的抗菌活性。

表2 TA、TAc和 TAd之间的比较
4.2 疏水性
抗菌肽的疏水性可以定义为抗菌肽序列中疏水性残基所占的比例。抗菌肽含有约 50%的疏水性氨基酸残基,其活性是亲水性残基和疏水性残基相互作用的结果。疏水性对抗菌肽活性的影响是通过改变肽链中Leu、Ile、Val数量进行测定的。研究发现:增加分子的疏水性,抗菌肽的抗菌活性和对哺乳类动物细胞的毒性同时增加。这是由于疏水性残基在抗菌肽插入细胞膜的过程中起到关键作用[27]。另外,由于疏水性残基的存在,肽链在溶液中可以通过疏水作用形成多聚体,增加了对真核细胞膜的亲和力。
未来的抗生素就是抗菌肽,因此对天然抗菌肽的序列进行设计和改造,使之具有更完美的抗菌活性、稳定性和更低的溶血活性,是人们追求的目标。抗菌肽设计是一个相当活跃的领域,多学科交叉在这里得到了充分体现。不同的研究者在设计不同抗菌肽的时候采用了不同的设计方法,虽然到目前为止,抗菌肽的设计取得了可喜可贺的成果,但是真正用于医疗和临床的抗菌肽少之有少。主要原因有:对抗菌肽的抗菌机理研究主要是通过研究抗菌肽与脂质体的相互作用得到的,因此抗菌肽的作用机理尚未完全研究清楚;多肽的合成周期较长,价格昂贵;多肽及蛋白质的空间构象研究进展较慢。未来,抗菌肽的设计仍然是研究的重点。生物信息学继续在这个过程中扮演着重要的角色,为抗菌肽的设计提供工具和数据库。近年来由于结构生物学的发展,相当数量的抗菌肽的三维结构被精确测定,使抗菌肽结构功能关系以及分子设计得以在三维结构上进行定性定量研究。相信在不久的将来,一定可以设计出理想的抗菌肽。
[1]Bonomo R A.Multiple Antibiotic-Resistant Bacteria in Long -Ter m-Care Facilities:An Emerging Problem in the Practice of in Factious Disease[J].Clin Infect Dis,2000,31(6):1414 -1422.
[2]肖建光,刘文生 .水产动物抗菌肽的研究进展[J].水利渔业,2005,25(1):4-5.
[3]Giangaspero A,SandriL,TossiA.Amphipathic Alpha Helical Antimicrobial Peptides[J].Eur J Biochem,2001,268(21):5589 -5600.
[4]ZasloffM.Antimicrobial Peptides ofMulticellular Organisms [J].Nature,2002,415(6870):389-395.
[5]Andreu D,Rivas L.An imal Antimicrobial Peptides:An Overview[J].Biopolymers,1998,47(6):415-433.
[6]李趣欢,张文军 .两亲α-螺旋抗菌肽的分子设计研究现状与进展[J].中国新药杂志,2005,14(9):1126-1132.
[7]Philippe Bulet,Reto Stocklin,Laure Menin.Anti-microbial Peptides:from Invertebrates to Vertebrates[J]. Immunological Reviews,2004,198:169-184.
[8]Song Yub Shin,Kyung-Soo Hahm.A Ahortα-helical Antimicrobial Peptidewith AntibacterialSelectivity[J]. Biotechnology,2004,26(9):735-739.
[9]SawaiMV,Waring AJ,Kearney WR,et al. Impact of single residue mutations on thestructure and function of ovispirin Pnovispirin ant imicrobial peptides[J].Protein Eng,2002,15(3): 225-232.
[10]Yanga ST,Shinb SY,Leea CW,et al.Selective cytotoxicity followingArg-to-Lys substitution in tritrpticin adopting a unique amphipathicturn structure[J].FEBS Letters,2003,540: 229-233.
[11]葛晓东,刘友生,杨艳丽,等 .人源 LL-37杀菌多肽的改建及原核细胞中表达[J].第三军医大学学报,2006,28(7): 636-639.
[12]Thorgerdur Sigurdardottir,Pia Andersson,Mina Davoudi,et al.In Silico Identification and Biological Evaluation of Antimicrobial PeptidesBased on Human Cathelicidin LL-37[J]. Antimicrobial Agents and Chemotherapy,2006,50(9):2983 -2989.
[13]Alessandro Tossi,Chiara Tarantino,Domenico Romeo. Design of Synthetic Antimicrobial Peptides Based on Sequence Analogy and Amphipathicity[J].Eur J Biochem,1997,250: 549:558.
[14]Tiozzo E,Rocco G,TossiA,et al.W ide-Spectrum Antibiotic Activity of Synthetic,Amphipathic Peptides[J].Biochem Biophys Res Commun,1998,249(1):202-206.
[15]Sung Yu Hong,Jong Eun Oh,Mi Yun Kwon,et al. Identification and Characterization ofNovelAntimicrobial Decapeptides Generated by Combinatorial Chemistry[J]. Antimicrobial Agents and Chemotherapy,1998,42(10):2534 -2541.
[16]FernandezLS,Kim HS,Choi EC,et al.Antibacterial Agents Based on Thecyclic D,L-Alpha-Peptide Architecture[J]. Nature,2001,412(6845):452-455.
[17]Yechiel Shai,Ziv Oren.From“Carpet”Mechanis m to De-novo Designed Diastereomeric Cell-Selective Antimicrobial Peptides[J].Peptides,2001,22(10):1629-1641.
[18]陈菲,严明,韦萍 .从 Granulysin抗菌机理出发合理设计抗菌肽[J].生物加工过程,2005,3(1):66-70.
[19]Ross Potter,Lisbeth Truelstrup Hansen,Tom A.Gill. Inhibition ofFoodborneBacteria by Native and Modified Protamine: Importance of Electrostatic Interactions[J].Inter J FoodMicrobial,2005,103:23-34.
[20]Sangita Roy,Prasanta KumarDas.AntibacterialHydrogels of Amino Acid-Based Cationic Amphiphiles[J].Biotecnilogy and Bioengineering,2008,100(4):756-764.
[21]Tanaka Munehiko.Modification of Functional Properties of Protamine and Polylysine[J].Foods&Food Ingred J Jpn,2000, 185:23-31.
[22]周家驹,雷静,谢桂荣 .药物设计中的模拟肽学[J].化学进展,2000,12(3):332-345.
[23]Shin SY,Lee SH,Yang ST,et al.Antibacterial Antitumor and Hemolytic Activities of Alpha-Helical Antibiotic Peptide, P18 and ItsAnalogs[J].J Pept Res,2001,58(6):504-514.
[24]Michael R Yeaman,Nannette Y Yount.Mechanisms of Antimicrobial Peptide Action and Resistance[J].Phar macological reviews,2003,55(1):27-55.
[25]Margitta Dathe,Heike Nikolenko,Jana Meyer,et al. Optimization of theAnt imicrobialActivityofMagainin Peptides by Modification of Charge[J].FEBSLetters,2001,501:146-150.
[26]Helena Hujakka,Jari Ratilainen,Timo Korjamo,et al. Synthesis and Antimicrobial Activity of the Symmetric Dimeric Form of Temporin A Based on 3-N,N-di(3-aminopropyl)amino Propanoic Acid as the BranchingUnit[J].Bioorganic&Medicinal Chemistry,2001,9:1601-1607.
[27]江龙法,谢慧,邬敏辰,杨海麟,等 .阳离子抗菌肽的作用机理及构效关系[J].中国医药工业杂志,2005,36(4):244-249.
Current status of molecular design of cation ic ant im icrobial peptides
WANGW ei,WANG L ian-jie*
(College of Science and Engineering,ShaanxiUniversity of Science&Technology,Xi’an 710021,China)
As the w ide ly use of trad itiona l antib iotics,m any p a thogenic m ic roorganism s exis t d rug res is tance to a lm os t a ll the current antib iotics.So,it is the comm on ta rge t of resea rche rs to look for a new gene ra tion ant im ic rob ia l d rug.The ca tionic ant im ic rob ia lp ep tide is exp ec ted to be a new gene ra tion of ant im ic rob ia l d rug w ith refe rence to the ir low re la tive m olecula r m ass,b road ant im ic rob ia l sp ec trum,s tab ility and sp ec ia l ant im ic rob ia l m echanism.The firs t p rob lem of exp loiting new ant im ic rob ia l d rugs is to inc rease its ac tivity,s tab ility and dec rease its toxic ity as we ll as cos t.M olecula r des ign of ant im ic rob ia l p ep tides is the key to the p rob lem.Dom es tic and fore ign resea rche rs have m ade a lot of contributions in the m olecula r des ign of ant im ic rob ia lp ep tides.Now,it ta lks about a ll the m e thods in m olecula r des ign of ant im ic rob ia lp ep tides,as we ll as the advantages and d isadvantages of each m e thod.A t the sam e t im e,the deve lopm ent foreg round of m olecula r des ign is d iscussed.
ant im ic rob ia lp ep tide;amp hip a thicα-he lica l;m olecula r des ign
TS201.2
A
1002-0306(2010)01-0442-05
自弗莱明发现青霉素以来,抗生素一直是人类治疗因病原微生物感染引起的疾病的有力武器,但是随着“传统抗生素”的大量使用,致病菌的耐药性逐渐增强,“抗生素时代”正逐渐走到尽头,目前最好的抗生素也逐渐失去效果,所以寻找新的抗菌药物已成为备受关注的焦点[1]。阳离子抗菌肽 (cationic ant imicrobial peptides)是一类对抗外界病原体感染的肽类活性物质,具有相对分子量小、抗菌谱广、热稳定性好和抗菌机理独特等优点[2]。但是天然的阳离子抗菌肽并非完美无缺,大部分天然抗菌肽存在抗菌活性不强、抗菌谱相对较窄、合成成本较高、部分抗菌肽对真核生物有一定的毒性、在具有对病原微生物高杀灭活性的同时往往伴随着对真核生物的溶血作用和对蛋白酶敏感等不足。因此如何提高其抗菌活性、扩大抗菌谱、降低合成成本和最大程度降低其毒性成为其分子设计的目的,也是目前抗菌肽药物开发的难点和希望所在。到目前为止,国内外的学者在阳离子抗菌肽设计方面做出了大量的工作,同时也取得了很多可喜的成果。
2009-02-20 *通讯联系人
王威(1985-),男,在读硕士,研究方向:抗菌肽设计。
