辽宁地区一脊髓小脑性共济失调大家系症状前患者的基因诊断研究
2010-08-21曹东华刘晓莉金春莲邱广斌
曹东华,王 谦,刘晓莉,金春莲,邱广斌
(1.中国人民解放军第二0二医院检验科,辽宁沈阳 110003;2.中国医科大学高职学院;3.中国医科大学基础医学院医学遗传学教研室)
辽宁地区一脊髓小脑性共济失调大家系症状前患者的基因诊断研究
曹东华1,3,王 谦2,刘晓莉3,金春莲3,邱广斌1*
(1.中国人民解放军第二0二医院检验科,辽宁沈阳 110003;2.中国医科大学高职学院;3.中国医科大学基础医学院医学遗传学教研室)
目的确定辽宁地区一脊髓小脑性共济失调大家系患者子女中目前健康的症状前患者。方法本家系已通过基因诊断的方法明确为SCA3型,所以提取本家系患者子女外周血DNA后,采用聚合酶链反应对SCA3基因三核昔酸重复片段进行扩增,并将PCR异常扩增片断测序,以明确异常扩增等位基因内重复拷贝数。结果44名患者子女中有8人SCA3基因三核苷酸重复片断在70-82次,超过了正常人SCA3基因内三核苷酸重复的次数。结论通过基因检测诊断出此8人为SCA3型脊髓小脑性共济失调症状前患者。
脊髓小脑性共济失调;三核苷酸重复;症状前患者
(Chin J Lab Diagn,2010,14:1420)
脊髓小脑性共济失调(spinocerebellar ataxia,SCA),是一大类由于遗传因素造成的进行性神经系统变性疾病,是一种单基因遗传病,多为常染色体显性遗传。其病理损害的主要部位是脊髓、脑干、小脑[1,2],临床上主要表现为共济失调,构音障碍、意向性震颤以及其它小脑症状。由于该病起病隐匿,临床症状互相重叠,基因检查是确诊和分型的唯一方法。我们于2009年在辽宁地区发现一脊髓小脑性共济失调大家系,经家系调查和基因诊断确定该家系为脊髓小脑性共济失调3型。由于该类疾病突变基因的相对稳定性,所以我们可以通过基因筛查发现症状前患者,并进行跟踪、临床随访,了解该病的发展过程。在本研究中,按照患者家属要求,我们对家系中患者的后代进行基因检测,现报告如下。
1 材料与方法
1.1 研究对象
本家系共5代198人(图1),前4代每代均有患者。由于年代久远,不能获得本家系中第1代的详细信息,因此不能明确致病基因来自Ⅰ1还是Ⅰ2。第2代也只能找到本身是患者且留下患者后代的分支,而第2代的正常分支则无从调查,因此本家系中第2代都是患者。本家系共有患者30人,目前存活13人。患者发病年龄最大39岁,最小20岁。前三代发病年龄都在35岁以上,第四代出现一例27岁发病患者和一例20岁发病患者。病史及体征基本相同,主要表现为行走不稳、步态蹒跚、饮水呛咳、语音低弱、构音障碍等共济失调症状和体征。我们通过基因检测,已确定本家为SCA3型(待发表)。
1.2 研究方法
1.2.1 前提条件 按照知情同意原则,基因测试必须获得受试者同意。向所有申请基因诊断者说明基因测试的目的、程序、可能的结果、目前能够检测的范围,并保证保守秘密、尊重其隐私权,如果受试人无法理解或为岁以下未成年人,则向其监护人、父母作出解释。
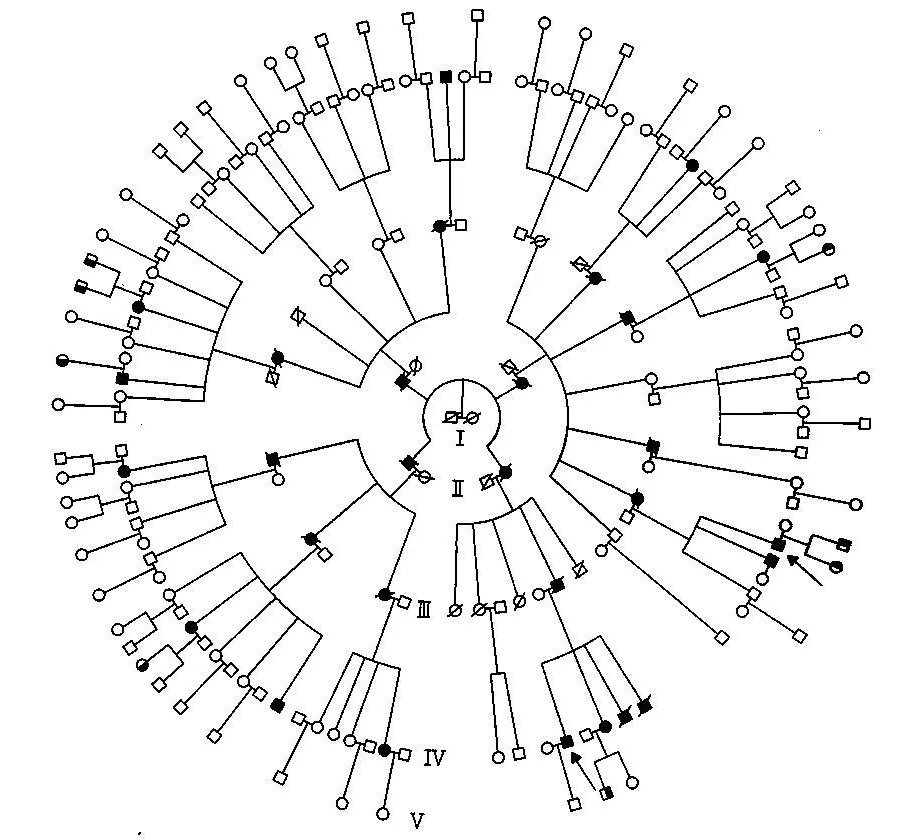
图1 家系图谱
1.2.2 基因组DNA提取 抽取本家系中所有患者子女中在世且未发病的成员共44人外周静脉血4 ml,枸椽酸钠抗凝,应用Tiangen血液DNA提取试剂盒提取基因组DNA作为模板。
1.2.3 SCA3三核苷酸重复片段PCR扩增 引物由上海生工合成,序列如下:Forward:5′-CTTTGATTCGTGAAACAATG-3′;Reverse 5′-GCCTTACCTAGATCACTCCC-3′。PCR 体系 :总体积 25 μ L,缓冲液 2.5 μ L,10×buffer 2.5 μ L,dNTP 2 μ L,上下游引物(20 μ mol/L)分别0.5 μ L,模板基因组DNA 约100 ng,TaKaLa Taq酶0.25 U,蒸馏水补齐,PCR反应在热循环仪上进行。循环参数如下:95℃预变性5 min后,进行35个循环:95℃变性45 s,55℃退火45 s,72℃延伸45 s,最后一次72℃延伸10 min。PCR产物2%琼脂糖凝胶电泳→发现异常条带后将异常条带纯化回收→与pMD18-T载体连接,转化于大肠杆菌JM109感受态细胞,经过蓝白筛选→取阳性克隆摇菌→菌液送金思特公司测序。
1.2.4 遗传咨询 检查结果书面通知受试者本人或其监护人,不得将测试结果告知第三者。向能够接受基因测试并可以理解者或其监护人解释的遗传方式、基因突变形式、简单的发病机理、目前的治疗现状,鼓励增强治病信心,学习必要的康复训练,加强锻炼。对未成年人强调不得告知阳性结果,不得歧视,保证其顺利成长。针对存在的外显率问题,慎重解释可能出现的后果。
2 结果
2.1 PCR产物检测结果 本家系患者子女中在世且未发病者共44人,其中8人SCA3基因CAG三核苷酸重复片段PCR扩增后有异常等位基因(图2)。
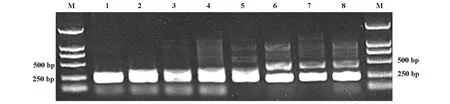
图2 8名无症状患者子女SCA3基因PCR扩增产物电泳图
2.2 异常等位基因CAG三核苷酸重复片段测序结果 8人SCA3基因 CAG三核苷酸重复70-82次(图3),证实此8人为症状前患者。
3 讨论
随着分子生物学技术水平的不断提高,医学遗传学、分子遗传学的发展,基因检查已逐渐走向临床,人们对脊髓小脑性共济失调这一大类疾病的认识以及发现率也不断提高。通过基因检测、分型有利地促进了的研究进展。按照分子遗传学分类,迄今为止共发现30种亚型。近年来大部分亚型的基因已被定位克隆,并且明确了三核苷酸重复序列动态突变,即致病基因内三核苷酸(CAG)n的拷贝数逐代增加的突变是主要致病原因。采用基因分型可以对患者作出准确的基因诊断,也正是由于直接而可靠的基因诊断技术的发展才有症状前诊断的出现。遗传性共济失调的突变机制的揭示使得可以在任何年龄阶段都能直接进行DNA检测,这必然带来复杂的与之相关的伦理、医学、社会问题。
国内外对其他神经遗传病的症状前诊断报道较多,但是关于SCA的症状前研究较少。国外学者对基因诊断操作提出了诸多相关的伦理问题并倾向于严格规范,并强调遗传咨询的重要性。症状前基因检测具有高度的敏感性和个人重要性,关系受试者本人的医疗保险、应聘就业、婚姻等,因而确保隐私是几乎所有受试者最优先考虑的问题。国内有关这方面的问题目前虽然还不十分突出,但是随着文化素质的提高、生活水平的改善,上述问题势必会同国外一样变得现实,应当引起重视[3,4]。因此我们在症状前患者的筛查时给予当事人或其监护人准确的遗传咨询十分重要。在进行遗传咨询时除了讲清SCA的遗传方式、临床表现、简要发病机理及预后外,还有必要讲明与测试有关的伦理、社会、法律、心理问题等[5]。
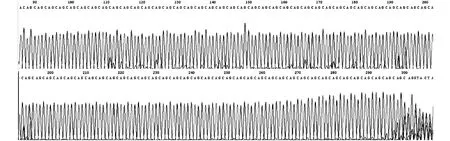
图3 症状前患者SCA3基因(CAG)n三核苷酸重复片段测序图
SCA3最早报道于葡萄牙人种,随后的发现显示这是一种全球性的疾病,在中国、日本及德国均为最常见的SCA类型[6],在中国的SCAs中约占一半左右。SCAs部分与三核苷酸重复序列有关,也有部分与点突变有关,还有部分未知。SCA3基因定位于14q24.3-q32.2[7],SCA3型患者是由于三核苷酸CAG重复次数增多所致,SCA3患者CAG三核苷酸重复数为53-86次,而正常人≤47次。介于47-53之间的中间类型有少量报道也显示存在异常临床表型[8]。本家系中在世的患者子女共44人,其中第3代4人,第4代22人,第5代16人。经基因检测分析,只有第5代中患者子女有8人的SCA3基因CAG三核苷酸重复在此范围内,可以推断此8人为症状前患者。第3代中患者子女仅存4人,而且年龄都在50岁以上,目前还没有发病,很可能是不携带致病基因,与基因检测结果相符。第4代中患者子女未发病者共22人,经基因检测此22人都不携带致病基因。此22人中大部分都在35岁以上,基本超过了本家系发病年龄,另有两人未到30岁,1人32岁,也不携带致病基因。第5代中患者子女共16人,年龄最大22岁,最小1岁,经基因检测,有8人携带致病基因。
有的遗传性疾病如肝豆状核变性,通过基因检测后发现症状前患者,给予一定的药物治病,可以预防该疾病的发生。但SCA缺乏有效的治疗手段,Miller等[9]报道可用RNA干扰的方法使基因沉默,但应用于临床尚有很长的距离。干细胞移植虽然可以暂时缓解SCA的症状,但并不能根治,而且医疗费用巨大,多数家庭承受不起。目前最为有效的控制SCA发生的手段就是应用产前诊断技术杜绝可能发病的后代出生。我们准备在此家系患者和症状前患者本人或爱人怀孕时,动员孕妇抽取羊水,以鉴定胎儿是否携带致病基因。如果胎儿携带致病基因则及时人工流产,以避免此家系再次出现患者,减轻其家族和社会的负担。
[1]Duenas A M,Goold R,Giunti P.Molecular pathogenesis of spinocerebellar ataxias[J].Brain,2006,129:1357.
[2]Netravathi M,Pal PK,Purushottam M,et al.Spinocerebellar ataxias types 1,2 and 3:age adjusted clinical severity of disease at presentation correlates with size of CAG repeat lengths[J].J Neurol Sci,2009,277(1-2):83.
[3]谢秋幼,李询桦,梁秀龄,等.脊髓小脑性共济失调的症状前诊断研究[J].脑与神经疾病杂志,2004,12(30):193.
[4]张小宁,雷 晶,马建华,等.脊髓小脑性共济失调症状前诊断的初步研究[J].脑与神经疾病杂志,2007,15(2):95.
[5]Tan EK,Ashizawa T.Genetic testing in spinocerebellar ataxia:defining a clinical role[J].Arch Neurol,2001,58(2):191.
[6]姜晓华,叶 蕾,傅 毅,等.遗传性脊髓小脑型共济失调一例家系SCA3基因突变研究[J].中华医学杂志,2005,85(12):848.
[7]谢秋幼,梁秀龄,李洵桦,等.我国南方汉族人脊髓小脑性共济失调不同基因亚型的频率分布[J].中华检验医学杂志,2004,27(9):555.
[8]van Alfen N,Sinke RJ,ZwartsMJ,et al.Intermediate CAG repeat lengths(53,54)forMJD/SCA3 are associated with an abnormal phenotype[J].Ann Neurol,2001,49:805.
[9]Miller V,Xia H,Marrs G,et al.Allele-specific silencing ofdominant disease genes[J].PNAS,2003,100:7195.
Gene diagnosis of asymptomatic carriers in a big spinocerebellar ataxias pedigree in LiaoNing
CAO Dong-hua,WANGQian,LIU Xiao-li,et al.(Department of Test,No.202Hospital of People'sLiberation Army,Shenyang110003,China)
ObjectiveTo identify the asymptomatic carrierswhose parents are affected in a big spinocerebellar ataxiaspedigree.Methods44 blood sampleswere collected from descendants of the patients in this Pedigree whichwas confirmed as SCA3 in previous study.The fragments of the SCA3 gene containing the CAG repeat region were amplified by means of polymerase chain reaction(PCR).The number of CAG repeatsin abnormal allele fragmentswas identifiedby using agarose gel electrophoresis(AGE)and DNA sequencing.ResultsThe CAG repeatnumbers in eight sampleswere in the range of 70-82,whichexceededthe normal range.ConclusionThe eight samples are SCA3 asymptomatic carriers by gene diagnosis.
Spinocerebellar ataxia;Trinucleotide repeat;Asymptomatic carriers
R745.1
A
1007-4287(2010)09-1420-03
*通讯作者
2010-02-08)
