表达序列标签的应用现状及分析方法研究
2010-07-25
(北京林业大学草地资源与生态实验室,北京 100083)
①表达序列标签 EST(Expressed Sequence Tag)是从一个随机选择的cDNA克隆进行5'端和3'端单一次测序获得的短的cDNA部分序列,代表一个完整基因的一小部分,在数据库中其长度一般从20~7 000 bp不等,平均长度为360±120 bp。EST作为表达基因所在区域的分子标签因编码DNA序列高度保守而具有自身的特殊性质,与来自非表达序列的标记(如 AFLP、RAPD、SSR等)相比更可能穿越家系与种的限制,因此EST标记在亲缘关系较远的物种间比较基因组连锁图和比较质量性状信息上是特别有用的。另外,由于EST来源于一定环境下一个组织总mRNA所构建的cDNA文库,因此EST也能说明该组织中各基因的表达水平。ESTs已经被广泛地应用于基因识别,研究发现ESTs的数目比GenBank中其他的核苷酸序列多,研究人员更容易在EST库中搜寻到新的基因[1]。由于EST测序只是测定部分序列,也不需要对克隆进行排序,因而完成EST测定所需要的人力、物力消耗与基因组测序和全长cDNA测序相比要少的多,具有经济和高效的特点。由于DNA测序技术的不断更新和大规模测序技术的出现,在DNA测序中逐步实现了工厂化和流水作业,因此测序费用大幅度降低[2]。
近年来,表达序列标签数据增长迅速。在GenBank102版本数据中,EST序列已经占用了2/3的记录[3]。美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)对EST进行了聚类分析,按基因划分EST,组成UniGene数据库。还有一些网站开发了基于Internet的EST延伸服务,如Labonweb网站的IRACE(http://www.labonweb.com),Biosino网站的BioEclone(http://bioinfo.bosino.org:9090/bioeclone.html)等[4-5]。
因此对EST的技术要求及应用进行归纳分析,有利于对研究对象分析不同基因的表达水平,为挖掘和克隆基因提供理论支撑。
1 EST特点及其应用
EST计划作为植物基因组计划的一个重要组成部分,已经在多种植物物种中开展起来。相关标记包括 EST-SSR、EST-PCR、EST-SNP、EST-AFLP、EST-RFLP等[6]。近年来,EST的应用已经深入到生物学的领域,其中表达序列标签微卫星(EST-SSR)技术的发展和应用较为普遍,根据SSR的来源可将其分为基因组SSR和EST-SSR[7]。EST-SSR标记狭义上是指位于EST序列上的或者基于EST序列开发的SSR标记,也被称为eSSR标记。
目前较为常用的核酸序列数据库有:美国国家信息中心的GenBank,欧洲分子生物学实验室的EMBL,日本国家数据库DDBJ,这3个数据库是收录范围最广并完全向公众开放的数据库,在它们中均含有EST子数据库dbEST。在核酸序列数据库中,EST的量要占65%以上[8]。
由于EST是功能基因的一部分,不同基因组间,基因编码区序列的保守性远远高于非编码区,与基因组SSR相比EST-SSR表现出较好的物种之间的可转移性[9-10]。作为一种新型分子标记,EST-SSR来自表达基因,因而除具备传统基因组来源的SSR标记的所有优势外,可能与基因功能表达具有直接或间接关系,从而强化了SSR标记在遗传研究中的应用[11]。
在种质资源遗传多样性方面,张鹏等[12]利用SRAP和EST-SSR分子标记对192份国内外芝麻Sesamum indicum进行分析。发现我国南部地区芝麻品种遗传多样性较中部和北部地区丰富。Eujayl等[13]利用EST-SSR等3种不同类型的微卫星标记对64个硬粒小麦Triticum aestivum品种的遗传多样性进行评价,表明EST-SSR可在硬粒小麦中揭示较高的多态性。
在基因连锁方面,利用分群分析法对多花黑麦草Lolium perenne抗叶斑病进行EST-CAPS标记,得到位点p56位于第5遗传连锁群,所处的基因为编码多花黑麦草天冬酰胺合成酶基因[14]。
在基因功能方面,郭久峰等建立沙冬青Ammopiptan thus mongolicus的cDNA文库并通过EST分析技术研究其抗逆机理,得到的313个已知功能的基因标签中抗逆相关的有48条[15]。
杨成君等[16]建立了药用植物人参Panax qinseng的EST-SSR标记。陈士林等[17]构建了西洋参P.quinquefolius的cDNA文库,经EST分析获得与水分胁迫相关的基因7个,与受伤诱导相关的基因2个,编码抗氧化酶相关的基因6个。并在根系的EST文库中发现抗病基因12个,62个 EST是其他物种尚未报道的新基因。佘玮等[18]以生长中期的苎麻Boehmeria nivea茎皮为材料构建cDNA文库,并进行EST分析,随即测序得到275个有效序列,约53.5%的 EST序列可能是未报道的新基因序列。
综上所述,EST为种质资源的保护利用和遗传育种工作提供科学依据,同时作为功能基因组研究的重要手段,在功能基因的开发与研究中也发挥重要作用。
2 EST在苜蓿中的研究
近几年,苜蓿Medicago sativa分子水平的研究有所深入,利用RAPD分子标记研究苜蓿种质资源遗传多样性[19]及其他相关基因的克隆序列分析等研究相对较多,如蒺蒺藜状苜蓿中MtERF-6基因的克隆及序列分析[20]。但EST的研究相对较少,闫娟等[21]利用EST-SSR标记分析了我国北部和中部地区天蓝苜蓿M.lupulina的遗传多样性和遗传结构,推测中等水平的遗传多样性和高度的居群间遗传分化主要受它的自交特性和分布方式影响。
在Genbank数据库中进行搜索,得出测序最多的 20个物种中,除经济类作物玉米Zea mays、水稻Oryza sativa、小麦等序列较多外,大部分物种为动物,蒺藜状苜蓿排在最后,序列条数为409 757。在表达序列标签数据库(dbEST)中进行搜索,测序最多的前20个物种中,没有和苜蓿相关的物种序列(总序列45 660 524条)。由以上数据可以看出,苜蓿基因的测序分析研究相对较少,只有蒺藜状苜蓿得到的EST较多,而紫花苜蓿和黄花苜蓿M.f alcata有待深入的研究。
3 EST获取及分析过程
3.1EST的获取过程构建生物某一发育阶段的cDNA文库,然后大规模、随机地挑选cDNA文库中的克隆或通过某种方法筛选cDNA中的某些克隆,最后对cDNA克隆的5'及3'进行测序,进而得到一个EST[22]。
3.2EST分析过程
3.2.1利用ESTs大规模分析基因表达水平 一般认为,组织和细胞分化依赖于基因特异性的时空表达,而生物体在某一时期的基因表达数量通常只占全部基因的15%[23]。
因为EST序列是从某种特定组织的cDNA文库中随机测序而得到的,所以可以利用未经标准化和差减杂交的cDNA文库EST分析特定组织的基因表达谱。标准化的cDNA文库和经过差减杂交的cDNA文库则不能反应基因表达的水平。
为研究癌症的分子机理,美国国家癌症研究所NCI的癌症基因组解析计划(Cancer Genome Anatomy Project,CGAP)构建了很多正常的或是癌症前期的和癌症后期的组织的cDNA文库,并进行了大规模的EST测序,其中大部分的文库未经标准化或差减杂交处理。CGAP网站提供了多种工具用以分析不同文库间基因表达的差异,如:Digital Gene Expression Displayer(DGED)和cDNA xProfiler。
3.2.2基因表达系列分析(Serial Analysis of Gene Expression,SAGE) 随着公用数据库中EST数据的急剧增加,基因表达研究可以利用数字化分析方法来实现[24-25],即从能够代表相应组织或器官基因表达情况的cDNA文库中获得大量EST,经过软件聚类拼接后依据代表基因的EST及其出现频率的信息进行基因表达分析。同样原理,也可以利用代表基因3'端表达信息的SAGE标签或近来出现的代表基因5'端信息的CAGE标签来进行。有学者把这种基于表达标签的基因表达水平定量分析方法称为数字化方法(digital method)或者数字化 Northern(digital Northern),而将传统的与cDNA克隆阵列和Oligo芯片杂交分析称为模拟方法(analog method)[26]。
Velculescu等[27]1995年提出基因表达系列分析是一种用于定量、高通量基因表达分析的实验方法。SAGE的原理就是分离每个转录本的特定位置的较短的单一的序列标签(约9~14个碱基对),这些短的序列被连接、克隆和测序,特定的序列标签的出现次数就反映了对应的基因的表达丰度。技术流程如图1。
3.2.3DNA微阵列或基因芯片的研究 随着ESTs数据的扩大,用ESTs文库制备的DNA芯片将使测序过程简化并有力促进功能基因组学研究[28]。
高密度寡核苷酸cDNA芯片或cDNA微阵列是一种新的大规模检测基因表达的技术,具有高通量分析的优点。在许多情况下,cDNA芯片的探针来源于3′EST[29],所以EST序列的分析有助于芯片探针的设计。以上几种方法比较,ESTs更适合大规模分析基因的表达水平(表 1)。
4 ESTS与基因预测
Adams等[30]提出的表达序列标签的概念标志着大规模cDNA测序时代的到来。虽然EST s序列数据相对不精确,精确度最高为97%[31],但实践证明EST技术可大大加速新基因的发现与研究。
由于EST来源于 cDNA,因此每一条 EST均代表了文库建立时所采样品特定发育时期和生理状态下的一个基因的部分序列。使用合适的比对参数,90%以上已经注释的基因都能在EST库中检测到[32]。ESTs可以作为其他基因预测算法的补充,因为它们对预测基因的交替剪切和3'非翻译区很有效。
4.1 测序方向的选择根据不同的试验目的选择不同的测序方向[33-34]:
1)5'端:5'端上游非翻译区较短且含有较多的调控信息。一般在寻找新基因或研究基因差异表达时用5'端EST较好,大部分EST计划都是选用5'端进行测序的,而且从5'端测序有利于将EST拼接成较长的基因序列。
2)3'端:3'端mRNA有一 20~200 bp的plyA结构,同时靠近plyA又有特异性的非编码区,所以从3'端测得EST含有编码的信息较少。但研究也表明[35],10%的mRNA 3'端有重复序列,这可以作为SSR标记;非编码区有品种的特异性,可以作为STS标记。
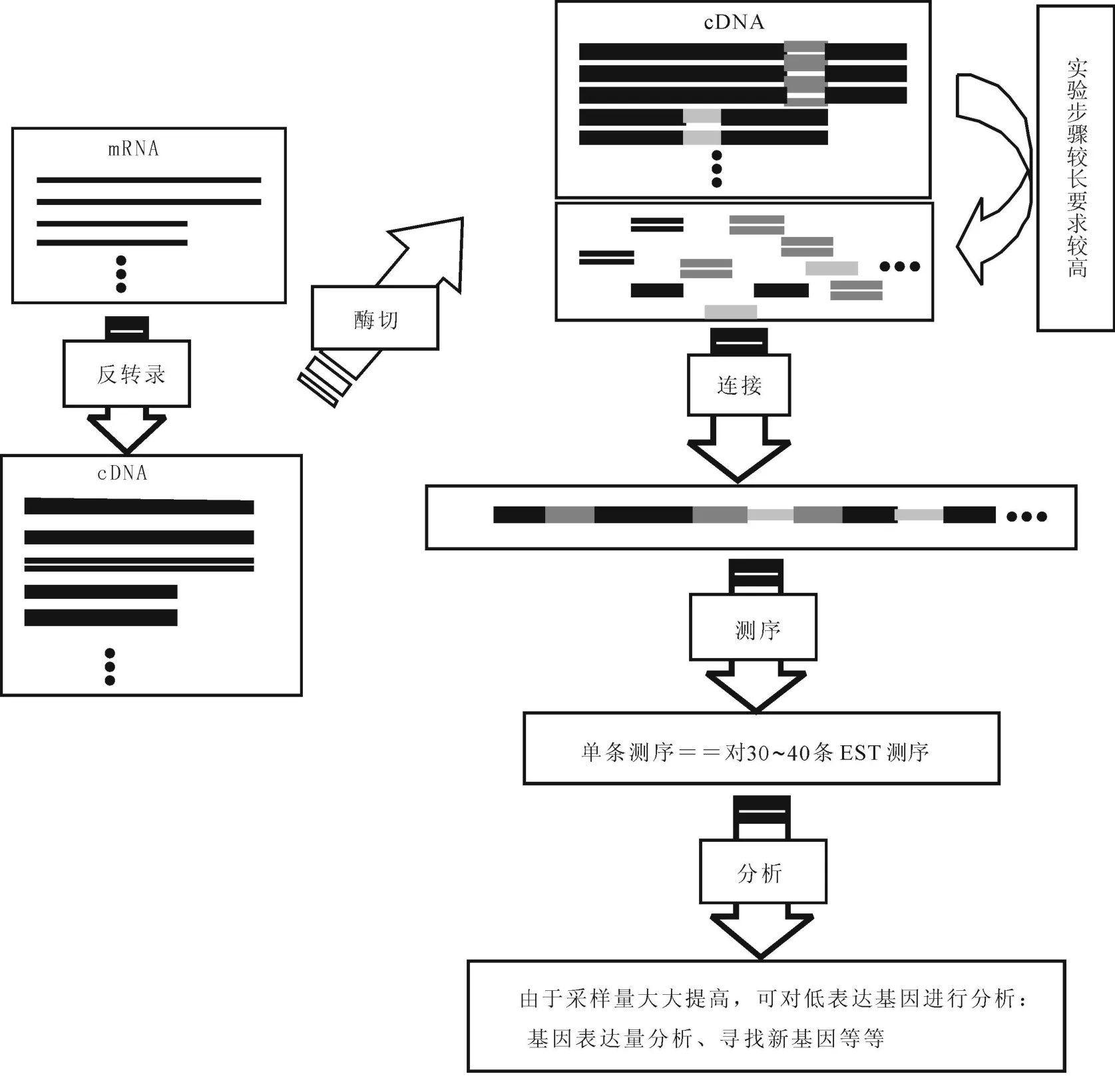
图1 SAGE技术流程

表1 几种大规模分析基因表达水平的方法比较
3)两端测序:获得更全面的信息。
4.2 序列前处理由于得到的序列包含一些不利因素,再聚类前要经过处理。主要涉及到:1)去除低质量的序列(Phred);2)应用BLAST、RepeatMasker或Crossmatch遮蔽数据组中不属于表达的基因的赝象序列(artifactual sequences),包括:载体序列(ftp://ncbi.nlm.nih.gov/reposi-tory/vector)、重复序列(RepBa,http://www.girinst.org)和污染序列(如核糖体RNA、细菌或其他物种的基因组DNA等);3)去除其中的镶嵌克隆,镶嵌克隆的识别包括Back-to-back poly(A)+tails、Linker-to-linker in middle of the sequence和Blastn/Blastx search;4)最后去除长度小于100 bp的序列。
4.3 聚类方法及ESTs聚类的数据库分析比较EST聚类(clustering)分析通过序列同源比较或其他注释信息,把属于同一基因的EST聚合成一簇,聚类的作用就是为了产生较长的一致性序列(consensus sequence),用于注释。降低数据的冗余,纠正错误数据可以用于检测选择性剪切[36]。
4.3.1聚类可分为不严格的和严格的聚类(loose and stringent clustering) loose clustering特点:产生的一致性序列比较长,含有同一基因不同的转录形式,如各种选择性剪接体。每一类中可能包含旁系同源基因(paralogous expressed gene)的转录本,序列的保真度低。如南非国家生物信息研究所(SANBI)的STACK[37]采用基于字的聚类算法(word-based clustering),省略了所有的比对过程,其核心在于识别并计算序列间有多少长度为n的字(word)能够匹配,代表性的算法有d2_cluster算法[38],是一种凝聚性的聚类算法。
而stringent clustering则产生的一致性序列比较短,表达基因ESTs数据的覆盖率低,因此所含有的同一基因的不同转录形式少,序列保真度高。它采用类似于BLAST和FASTA的序列比对的算法,通过寻找序列间的局部相似性来判断两序列是否具有重叠片段或连续的匹配,并据此聚类。如NCBI的Unigene[39](此系统同时还利用一些注释信息,如EST序列的克隆号)以及美国基因组研究所(TIGR)的 Gene Indices[40](TGICL聚类,适用于大规模EST序列的快速聚类,并可进行连锁分析)。
4.3.2ESTs聚类的主要数据库
1)UniGene(http://www.ncbi.nlm.nih.gov/UniGene):UniGene Clustering方法由美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)发展而来。该方法使用MEGABLAST程序[41]对序列进行同源比较,采用的聚类阈值为序列间至少有100个碱基的重叠区,并且占70%以上的重叠区域的碱基同源性大于96%,依据该阈值先对已注释的基因聚类成簇,再根据EST与EST及EST与初始基因簇之间的序列同源性进一步进行聚类,由此产生的基因簇包括同一基因的不同剪接形式。
2)TIGR Gene Indices(http://www.tigr.org/tdb/tgi/):目前,根据不同的研究目的发展了多种EST聚类分析方法,其中被广泛使用的有美国基因组研究所发展而来的TIGR-ASSEMBLER方法[42]。该方法借助FASTA程序[43]对序列进行两两比较,再根据同源比较结果用 TIGR-ASSEMBLER工具对相关序列进行拼接,把重叠区超过40个碱基,且该区域的碱基同源性大于95%的序列合并成一簇。TIGR利用这个方法对来源21个物种的5 358 611条EST进行了聚类分析,分别建立了各个物种的基因索引(TIGR Gene Indices)[44]。
3)STACK:南非国家生物信息研究院(South African National Bioinformatics Institute,SANBI)的STACK-PACK方法(http://www.sanbi.ac.Za/Dbases.html),其主要特点是根据不同的组织来源先把EST分类,再根据重叠区超过150个碱基,且重叠区域的碱基同源性大于96%的聚类阈值,用d2-cluster程序[45]对各类EST分别聚类。用STACK-PACK分析结果建立的STACK数据库可用来进行SNPs检测和基因特异性表达的研究[46]。
目前有学者已经提出了一种基于样本间关系的新聚类方法,即从基因表达数据中通过pearson相关系数获得样本间的关系,并用网络的方法表示这种关系,通过该网络的空间结构特征来提取样本间的关系特征,并在这种关系特征空间中进行样本的聚类[47]。
4.3.3基于BLAST和FASTA的脚本(BLASTN and FASTA-based scripts) 在 EST研究中,使用最多的方法就是序列相似性比较,以此来确定EST的功能。BLAST(Basic Local A-lignment Search Tool)是应用较广的工具软件之一,为同源分析的软件包,包括 BLASTN、BLASTP 、TBLASTN 、TBLASTX 、BLASTX 5 个软件[48]。
4.3.43个数据库的比较分析
1)UniGene:结合有指导的和无指导的方法,而且在聚类过程中使用了不同水平的严格度,聚类的算法为megablast,数据库不产生一致性序列。
2)TIGRGene Index:用的是有严格的和有指导的聚类方法,聚类的算法为类似于BLAST和FASTA的FLAST,该法得到的一致性序列较短,交替剪切得到的不同的基因属于不同的索引。
3)STACK:用不严格的和无指导的聚类方法,聚类的算法为d2_cluster,产生较长的一致性序列,同一索引中含有不同的剪切方法得到的基因。
4.4EST接拼由于高中比率重复序列的存在、克隆文库时产生的无可回避的间隙、基因的多态性以及测序技术或实验室一些人为因素引起的错误等的存在,要想把序列拼接正确是非常困难的[49]。Cluster的连接:利用cDNA克隆的信息和5',3'端Reads的信息,不同的Cluster可以连接在一起。常用的拼接软件为Phrap。Phrap常与phred、cross-match、consed组成一个软件包,通常用的是 perl写的脚本程序 PhredPhrap。Feng Liang 等比较了 Phrap、CAP3、T IGR Assembler,认为CAP3是最佳的软件[50-51]。
4.5 基因注释及功能分类注释的过程包括序列比对寻找同源基因和蛋白结构功能域的搜索。
4.5.1注释 EST数据注释通常先做blastx比对,将所有未知的EST序列按理论上的6种阅读框翻译为蛋白质序列,在非冗余的蛋白序列数据库(Non-redundantprotein Sequence Database,NR)中搜索同源序列,它提供所有可能的翻译结果的比对,并且对每个比对结果进行综合的显著性分析。在blastx中不能匹配上的核酸序列可以继续通过blastn搜索相应的核苷酸序列数据库(Nucleotide Sequence Database,NT)。如果要进一步验证BLAST结果,或者更详细了解蛋白序列信息,可以通过InterPro来补充和完善。同样通过BLAST无法注释的序列,可以进一步通过InterPro数据库,搜索序列中可能含有的蛋白功能结构域(domain)、模体(motif)等信息[52]。
局部序列比对工具BLAST是最常用的相似性检索工具[53-54]。用户可以登录NCBI网站(http://www.ncbi.nlm.nih.gov/BLAST/)进行检索,也可在其他网站上进行检索,还可以下载于本地运行(ffp://ftp.ncbi.nih.gov/)。
4.5.2基因功能分类 基因功能的分类可分为手工分类和计算机批量处理。
其中手工分类的大部分以Adams等[55]提出的分类体系为标准。而计算机批量处理则是利用标准基因词汇体系Gene Ontology进行近似的分类。其结果将会发现与已知功能的蛋白具有高度同源性的已知基因(known genes),与未知功能的蛋白具有高度同源性的未知功能基因(unknown genes)和仅有很低同源性或没有同源蛋白的序列,记为新基因(novel genes)[56]。GO注释分为3个层次,分别说明基因产物执行哪种分子功能、参与哪个生理过程以及定位于哪个细胞部位[57]。
4.6 后续分析EST方法的优点在于它不需要很多关于目的基因的假设,可为后续的研究提供大量基因资源信息[58]。
所谓后续分析即EST通过以上聚类接拼,将基因功能分类后,进行比较基因组学分析、基因表达谱分析、新基因研究、基因可变剪切分析、实验验证(MicroArray、GeneChip、RTPCR、Northen bloting)[59]。
用EST取代对cDNA全长的筛选、基因组序列的鉴定等繁琐的实验操作,可大大地提高分离基因的效率。将所获EST用生物信息学方法与各公共数据库中已知序列进行比较,可迅速而准确地确定基因功能。由于在构建cDNA文库时要尽可能地选用全长cDNA,所以一旦发现有价值的EST,就可以找到对应的克隆,获得的全长cDNA可以直接用于如转基因等的研究。
5 EST的问题与展望
用于构建的普通cDNA文库进行测序时,由于EST测序时克隆的挑选是随机的,高峰度表达基因引起mRNA的表达水平高而被反复测序;相反,一些峰度较低的基因需要测定上万个克隆才有可能被挑选测序。因此,对于为寻找新基因或研究基因差异表达而言,用这样的cDNA文库进行测序,一方面稀有基因容易遗漏,及EST很短,没有给出完整的表达序列,相对较低丰度表达基因不易获得[60]。另外,由于只是一轮测序结果,出错率达2%~5%。有时有载体序列和核外mRNA来源的cDNA污染或是基因DNA的污染会对实验造成一定影响。镶嵌克隆的出现以及序列的冗余都会导致所需要处理的数据量很大。
利用EST方法进行发现、分离基因的研究,不仅是人类基因组研究的热点,而且是植物基因组研究的重要内容[61-62]。这将为人们更好地了解功能基因在不同组织中的表达提供分子生物学依据,从而为将来在分子水平调控生物的生长、发育、抗性和代谢规律打下理论基础,提供极有价值的资源。
[1]Boguski M S,Tolstoshev C M,Bassett D E,et al.Gene discovery in dbEST[J].Science,1994,30(9):4.
[2]Bhattramakki D,Chhabra A K,Hart G E,et al.An Integrated SSR and RELP Linkage M ap of Sorghum Bicolor Moench[J].Genome,2000,43(6):988-1002.
[3]李衍达,孙之荣.基因和蛋白质分析的实用指南[M].北京:清华大学出版社,2000.
[4]郝柏林,张淑誉.生物信息学手册[M].上海:上海科学技术出版社,2000.
[5]李越中,闫章才,高培基.基因组研究与生物信息学[M].济南:山东大学出版社,2001.
[6]陈全求,詹先进,蓝家样,等.EST分子标记开发研究进展[J].农业生物技术科学,2008,24(9):72-77.
[7]Chen C,Zhou P,Choiya,et al.Mining and characterizing microsatellites from citrus ESTs[J].TAG theoretical and applied genetics,2006,112(7):1248-1257.
[8]Leipe D D.Genome and DNA sequence database[J].Curr Opin GenDevel,1996,6(6):686-691.
[9]Hanai L R,Campos T,Camargo L E,et al.Development,characterization,and comparative analysis of polymorphism at common bean SSR loci isolated from genic and genomic sources[J].Genome,2007,50(3):266-277.
[10]Ellis J R,Pashley CH,Burke J M,et al.High genetic diversity in a rare and endangered sunflower as compared to a common congener[J].Mol Ecol.,2006,15(9):2345-2355.
[11]吴曼颖,刘昆玉,方芳,等.EST-SSR标记的开发及在果树上的应用研究进展[J].江西农业学报,2009,21(5):59-62.
[12]张鹏,张海洋,郭旺珍,等.以SRAP和 EST-SSR标记分析芝麻种质资源的遗传多样性[J].作物学报,2007,33(10):1696-1702.
[13]忻雅,崔海瑞.植物表达序列标签(EST)标记及其应用研究进展[J].生物学通报,2004,39(8):4-6.
[14]丁成龙,沈益新,顾共如,等.分群分析法获得与多花黑麦草抗叶斑病基因连锁的 EST-CAPS标记[J].草地学报,2006,14(1):9-13.
[15]郭九峰,孙国琴,沈传进,等.沙冬青 cDNA文库的构建和EST分析[J].华北农学报,2007,22(4):37-41.
[16]杨成君,王军,穆立蔷,等.人参EST-SSR标记的建立[J].农业生物技术学报,2008,16(1):114-120.
[17]陈士林,孙永巧,宋经元,等.西洋参 cDNA文库构建及表达序列标签(EST)分析[J].药学学报,2008,43(6):657-663.
[18]佘玮,邢虎成,秦占军,等.苎麻茎皮表达序列标签(ESTs)分析[J].热带作物学报,2008,29(2):657-663.
[19]蒿若超,张月学,唐凤兰,等.利用 RAPD分子标记研究苜蓿种质资源遗传多样性[J].草业科学,2007,24(8):69-73.
[20]连瑞丽,李宇伟,赵德刚,等.蒺藜状苜蓿中MtERF-6基因的克隆及序列分析[J].草业科学,2006,23(9):82-87.
[21]闫娟,楚海家,王恒昌,等.用 EST-SSR标记分析中国北部和中部地区天蓝苜蓿的遗传多样性和遗传结构[J].生物多样性,2008,16(3):263-270.
[22]刘伟,邵菁,庞宏,等.大规模筛选表达序列标签(EST)方法的改进[J].安徽农业科学,2007,35(24):7410-7411.
[23]Happe T,Kaminski A.Diferential regulation of the Fe-hydrogenase during adaptation in the green alga Chlamydomonas reinhardtii[J].European Journal of Biochemistry,2002,269(3):1022-1032.
[24]Rob M E,Alia B K,Olivier P,et al.Large-scale statistical analyses of rice ests reveal correlated patterns of gene expression[J].Genome Research,1999,9(10):950-959.
[25]Velculescu V E,Zhang L,Vogelstein B,etal.SAGESerial analysis of gene expression[J].Science,1995,270(10):484-487.
[26]赵光耀,孔秀英,贾继增,等.粗山羊草幼苗和根全长cDNA文库构建及其EST注释与比较分析[J].中国农业科学,2007,40(7):1331-1336.
[27]Audic S,Claverie J M.The significance of digital gene expression profiles[J].Genome Research,1997,7(10):986-995.
[28]李红,卢孟柱,蒋湘宁,等.表达序列标签(EST)分析及其在林木研究中的应用[J].林业科学研究,2004,17(6):804-809.
[29]Kleinbaum L A,Duggan C,Ferreira E,et al.Human chromosomal localization,tissue/tumor expression,and regulatory function of the ets family gene EHF[J].Biochemical and Biophysical Research Communications,1999,264(1):119-126.
[30]Adams M D,Kelley J M,Gocayne J D,et al.Complementary DNA sequencing:expressed sequence tags and human genome project[J].Science,1991,21(6)252:1651-1656.
[31]Bailey L C,Jrsearls D B,Dverton G C.Analysis of EST-driven gene annotation in human genomic sequence[J].Genome Research,1998,8(4):362-376.
[32]Hillier L,Lennon G,Becker M,et al.Generation and analysis of 280,000 human expressed sequence tags[J].Genome Research,1996(6):807-828.
[33]Hatey F,Tosser-Klopp G,Clouscard-martinato C,et al.Expressed sequenced tags for genes:a review[J].Genet Sel Evol,1998,30(5):521-541.
[34]Yammanoto K,Sasaki T.Large-scale EST sequencing in rice[J].Plant Molecular Biology,1997,35(1):135-144.
[35]张建成,王传堂,杨新道,等.SSR和STS标记在花生栽培品种鉴定中的应用研究[J].植物遗传资源学,2006,7(2):215-219.
[36]Hide W,Miller R,Ptitsyn A,etal.EST Clustering tutorial[C].Heidelberg:ISMB Germany,1999.
[37]Christoffels A,van Gelder A,Greyling G,etal.STACK:Sequence tag alignment and consensus knowledgebase[J].Nucleic Acids Res,2001,29(1):234-238.
[38]Bmke J,Davison D,Tlide W.d2_cluster:a validated method for clustering EST and full-length cDNAsequences[J].Genome Res,1999,9(11):1135-1142.
[39]Schuler G D.Pieces of the puzzle:expressed sequence tags and the catalog of human genes[J].J Mol Med,1997,75(10):694-698.
[40]Lee Y,Tsai J,Sunkara S,et al.The TIGR Gene Indices:clustering and assembling EST and known genes and integration with eukaryotic genomes[J].Nucleic Acids Res,2005,33(Database issue):71-74.
[41]Zhang Z,Schwartz S,Wagner L,et al.A greedy algorithm for aligning DNA sequences[J].J Comput Biol,2000,7(1-2):203-214.
[42]Sutton G,White O,Adams M D,et al.TIGR Assembler:A new tool for assembling large shotgun sequencing projects[J].Genome Sci Techno,1995(1):9-18.
[43]Pearson W R,Lipman D J.Improved tools for biological sequence comparison[J].Proc Natl Acad Sci USA,1988,85(8):2444-2448.
[44]Quackenbush J,Cho J,Lee D,et al.The TIGR Gene Indices:Analysis of gene transcript sequences in highly sampled eukaryotic species[J].Nucleic Acids Res,2001,29(1):159-164.
[45]Burke J,Davison D,Hide W.d2_cluster:a valida-ted method for clustering EST and full length cDNA sequences[J].Genome Res,1999(9):1135-1142.
[46]Miller R T,Christoffels A G,Gopalakrishnan C,et al.A comprehensive approach to clustering of expressed human gene sequence:The sequence tag alignment and consensus knowledge base[J].Genome Res,1999(9):1143-1155.
[47]王文俊,张军英.一种新的基因表达数据聚类方法[J].西安电子科技大学学报,2009,36(3):502-505,534.
[48]夏云,雷二庆,王槐春.Internet实用技术与生物医学应用[M].北京:军事医学出版社,1997:341-354.
[49]Green P.Against a whole-genome shotgun[J].Genome Res,1997,7(5):410-417.
[50]Jacek B,Marek F.Assembling the SARS-CoV genome-new method based on graph theoretical approach[J].Acta Biochimica Polonica,2004,51(4):983-993.
[51]Carvalho C M L,Melo E P,Cabral J M S,etal.A steady-state fluorescence study of cutinase microencapsulated in AOT reversed micelles at optimal stabilityconditions[J].Journal of Biotechnology,1998,21(8):673-681.
[52]刘稳升,吴忠道.表达序列标签大规模序列分析策略及方法[J].国际医学寄生虫病杂志,2007,34(3):139-145.
[53]Altschul S F,Madden T L,Schafer A A,et al.Gapped BLASI1 and PSI-BLAST:a new generation of protein database search programs[J].Nucleic Acids Res,1997,25(17):3389-3402.
[54]Altschul S F,Gish W,Miller W,etal.Basic local alignment search too1[J].J Mol Biol.,1990,215(3):403-410.
[55]Adams M D,Kerlavage A R,Fleischmann R D,et al.Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence[J].Nature,1995,377:3-174.
[56]钱骏,董利.表达序列标签数据库搜索鉴定小鼠UBAP1基因及其数字化表达分析[J].生物化学与生物物理进展,2002,29(2):323-327.
[57]赵光耀,孔秀英,贾继增,等.粗山羊草(Ae.tauschii)幼苗和根全长cDNA文库构建及其EST注释与比较分析[J].中国农业科学,2007,40(7):1331-1336.
[58]孙亮先,袁建军.EST技术在植物基因克隆和基因表达谱研究中的应用[J].泉州师范学院学报,2003,21(4):63-67.
[59]崔佳欣,孟军,朱荣胜,等.大豆表达序列标签(ESTs)研究进展[J].东北农业大学学报,2009,40(2):123-126.
[60]张新.表达序列标签(EST)的研究现状[J].黑龙江医学,2008,32(9):676-678.
[61]Rounsley S,Linx K K.Large scale sequencing of plant genome[J].Curr Opin Plant Biol,1998,1(2):136-141.
[62]Sasaki T.The rice genome project in Japan[J].Proc Natl Acad Sci USA,1998,95(5):2027-2028.
