线粒体脑肌病一例
2010-07-05毋凡,陈聪
毋 凡,陈 聪
(1.中国人民解放军第187中心医院放射科,海南 海口 571159;2.中国人民解放军第75608部队卫生队,海南 海口 571159)
线粒体脑肌病(Mitochondrial encephalomypathy)是以线粒体的形态和功能异常而命名的一组疾病,因临床症状复杂,影像学表现不具有特异性,极易误诊,现报告1例被误诊为病毒性脑炎的病例。
1 病例简介
患者,男性,19岁。2009年12月21日入院,见无明显诱因下间歇性头痛,乏力、听力下降5 d,问话反应迟钝。2010年6月1日入院后查脑积液常规:白细胞(WBC)7.5×109/L,红细胞(RBC)4.3×109/L。脑脊液生化:葡萄糖(GLU)3.0 mmol/L,血氯(Cl)114.1 mmol/L,血乳酸4.36 mmol/L。脑电图轻-中度异常,脑膜刺激征阴性。肌肉活检确诊线粒体脑肌病。
2 影像学表现
MRI示:右侧枕叶区见大片状长T1长T2信号,T2FLAIR序列呈高信号,以灰质受累为主,受累脑沟变浅,脑回增粗,Gd-DTPA增强后未见异常强化(见图1)。6个月后MRI示:右侧枕叶病灶明显好转,左侧颞叶区见大片状长T1长T2信号,T2FLAIR序列呈高信号,Gd-DTPA增强后未见异常强化(见图2)。治疗半月后再次复查,左侧颞叶区病灶好转,未见新病灶(见图3)。
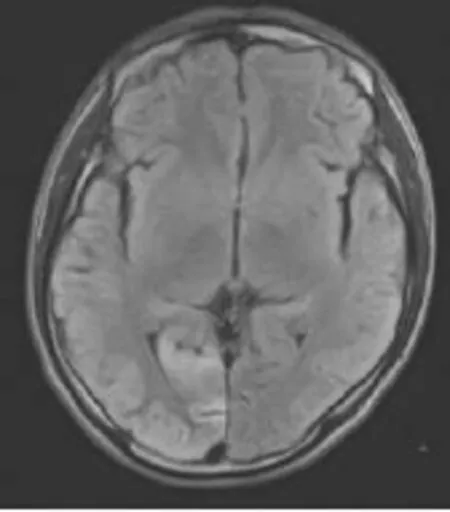
图1 T2FLAIR序列于右枕叶见脑回状高信号影,以皮质受累为主,受累脑沟变浅,脑回增粗。

图2 (a-d)6个月后,患者右侧枕叶病灶基本消失,于左颞叶见不规则片状稍长T1长T2信号影,T2FLAIR序列呈高信号,边界欠清,病变仍以灰质受累为主,Gd-DTPA增强后病变区未见明显强化。
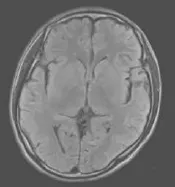
图3 治疗半个月后再次复查,左侧颞叶区病灶好转。
3 讨论
线粒体脑肌病是以线粒体DNA基因缺陷导致的多系统疾病,以脑和肌肉受累最多,它包含一组病,如Kearns-Sayre综合征、弥散性进行性脑灰质变性(Alpers)综合征、线粒体脑肌病伴乳酸血症及卒中样发作(MELAS)等。多数为散发,也可由母系传递之隐性遗传,3-5岁后发病。临床症状复杂,表现多样,神经系统影像学以出现坏死性病变为特征,并表现为脑组织内多发性软化灶。文献认为线粒体脑肌病不是罕见病,多是由医师对其缺乏认识或检查方法限制,造成很多病例误诊[1-2]。
线粒体脑肌病的MRI表现无明显特征性[3-5],表现多样:①以大脑灰质损害为主,可见皮质异常信号,多累及半球后部颞顶枕叶,近似于脑梗塞体表改变,但病变的范围不是血管分布范围,以MELAS为多见;Alpers病亦常见到上述征象。②对称性双侧基底节、丘脑、脑干等异常信号。③灰质和白质散在异常信号,见于Kearns-Sayre综合征;白质病变多侵犯较新的周围白质,即皮质下和三角区后部白质[6]。
本例患者临床神经症状明显,病情反复,MRI检查时其病灶逐渐发展,由枕叶到颞叶,以皮质灰质受累为主,病变不按血管分布,有自愈倾向,增强扫描后无强化,表明病变无明显血脑屏障破坏。本例被误诊为病毒性脑炎,经抗炎治疗后效果不佳。后详细询问其母亲有该病史,结合家族史及肌肉活检,确诊为线粒体脑肌病。
总之,线粒体脑肌病的MRI表现多样,影像学检查不具有特异性,但当患者有难以解释的神经、肌肉等系统症状,影像学表现幕上病灶,应仔细观察病变的信号特点、分布和其他病变的差别,结合临床病史,多应考虑到此病,以减小误诊率。
[1] 陈清棠,吴丽娟,伍期专,等.原发性线粒体肌病与脑肌病(附53例报告)[J].中国神经精神疾病杂志,1994,20(1):16-18.
[2] Servidci S,Zeviani M,Manfredi G,et al.Dominanty inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA:clinical,morphologic,and biochemical studies[J].Neurology,1991,41:1053.
[3] 马梦华,王海平,韩德昌,等.线粒体脑肌病的MRI诊断价值[J].临床放射学杂志,2007,26(1):14-17.
[4] 王霄英,肖江喜,学 祥,等.儿童线粒体脑肌病的MRI表现[J].中国影像学影像技术,2001,17(4):314-316.
[5] 杨 波,黎桂平,刘梦雨,等.线粒体脑肌病表现特征及诊断价值[J].放射学实践,2008,23(4):368-372.
[6] 邢 妩,王小宜,廖伟华,等.线粒体脑肌病的MR诊断[J].中国介入影响与治疗,2010,7(1):35-37.
