Roles of Smad3 and Smad7 in rat pancreatic stellate cells activated by transforming growth factor-beta 1
2010-06-29ZhuYinQianQuanPengZhengWeiZhangLongAnZhouandYiMiao
Zhu-Yin Qian, Quan Peng, Zheng-Wei Zhang, Long-An Zhou and Yi Miao
Nanjing, China
Original Article / Pancreas
Roles of Smad3 and Smad7 in rat pancreatic stellate cells activated by transforming growth factor-beta 1
Zhu-Yin Qian, Quan Peng, Zheng-Wei Zhang, Long-An Zhou and Yi Miao
Nanjing, China
BACKGROUND:Pancreatic stellate cells (PSCs) play a major role in promoting pancreatic fibrosis. Transforming growth factor beta 1 (TGF-β1) is a critical mediator of this process. This study aimed to determine the expression of the Smad3 and Smad7 genes in the process of PSC activation, and explore the mechanisms of chronic pancreatitis.
METHODS:The expressions of Smad3 and Smad7 in PSCs before and after TGF-β1 treatment were detected by reverse transcription-polymerase chain reaction and Western blotting analysis. Smad3 expression was detected in PSCs after treatment with 5 ng/ml of TGF-β1 for 24 hours.
RESULTS:Smad7 expression was decreased in TGF-β1-activated PSCs (P<0.05) in a dose-dependent manner. When TGF-β1 concentration reached 10 ng/ml, the expression of p-Smad3, Smad3, and Smad7 was inhibited (P<0.05).
CONCLUSIONS:TGF-β1 promotes the expression of Smad3 and inhibits the expression of Smad7 during the activation of PSCs. In contrast, high-dose TGF-β1 downregulates the expression of Smad3 in completely activated PSCs.
(Hepatobiliary Pancreat Dis Int 2010; 9: 531-536)
pancreatic stellate cell; transforming growth factor beta 1; chronic pancreatitis; Smad3; Smad7
Introduction
In 1982, Watari et al[1]first reported vitamin A-storing cells in rat pancreas. In 1990, Ikejiri[2]named these cells pancreatic stellate cells (PSCs), then Bachem et al[3-5]successfully isolated PSCs from human and rat pancreas. They reported that PSCs are capable of transforming from a quiescent phenotype into myofibroblast-like cells that produce large amounts of extracellular matrix (ECM), including the proteins collagen typesiand III, fibronectin, and laminin. Recently, studies supported byin vitroandin vivodata have suggested that PSCs are involved in the development of pancreatic fibrosis,[6-10]and PSCs respond to various proinflammatory and profibrogenic cytokines. Transforming growth factor β1 (TGF-β1) is a major profibrogenic cytokine. The intracellular signaling of TGF-β1 is mediated and modulated primarily by Smaand Mad-related proteins (Smads). Studies have revealed that TGF-β1 stimulates PSC activation in a Smad2-dependent manner, while Smad3 is required for TGF-β1-induced growth inhibition. Smad7, a negative regulator of TGF-β signaling, might act as a transcriptional inhibitor of PSC activation. However, there are little experimental data to support this hypothesis. In this study, we investigated changes in the expression of Smad3 and Smad7 in PSCs transforming from the quiescent stage to the completely activated stage under the influence of TGF-β1.
Methods
Reagents
Collagenase P was from Roche Molecular Biochemicals (Mannheim, Germany). DNaseiand protease were from Sigma (St. Louis, MO, USA). Nycodenz was from Axis-Shield (Oslo, Norway). TGF-β1, mouse anti-rat desmin, and α-SMA monoclonal antibodies were from BosterWuhan Biological Technology Ltd. (Wuhan, China). Ultrasensitive S-P and DAB kits were from Maixin Biological Co. (Fuzhou, China). Dulbecco's modified Eagle's medium (DMEM) was from Gibco-BRL (Gaithersburg, MD, USA). FCS was from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China). Goat anti-human p-Smad2/3, antihuman Smad7, rabbit anti-human Smad3, horseradish peroxidase (HRP)-rabbit anti-goat, and HRP-donkey anti-rabbit polyclonal antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Penicillinstreptomycin and Trizol were from Invitrogen (Carlsbad, CA, USA). The RT-PCR kit was from Shanghai Sangon Biological Engineering Technology & Service Co., Ltd. (Shanghai, China). The ECL kit was from Pierce Biotechnology (Rockford, IL., USA).
PSC isolation, culture and treatments
Using a simple collagenase perfusion method through abdominal aorta cannulation to isolate rat PSCs, PSCs were identified by morphology, cytoplasmic lipid droplets, and immunocytochemical staining for desmin, glial fibrillary acidic protein and α-smooth muscle actin (α-SMA).[11]Briefly, male Sprague-Dawley rats weighing 300-400 g were bought from Shanghai Laboratory Animals Co. Ltd. (Shanghai China). The pancreas was digested with a mixture of collagenase P, pronase and deoxyribonuclease in Gey's balanced salt solution. The digested tissue was pipetted through narrow orifices and filtered through 200-mm mesh. The resulting cell suspension was centrifuged in a 15% Nycodenz gradient at 1400 g for 20 minutes. PSCs were then separated into a hazy band just above the interface of the Nycodenz solution and the aqueous buffer. The isolated PSCs were cultured in DMEM supplemented with 10% FCS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin in a 5% CO2atmosphere at 37 ℃. The viable cells were determined by 0.4% trypan blue staining analysis, and the purity of isolated cells was confirmed to be >90% by observing their cytoplasmic droplets with vitamin A autofluorescence. PSCs were divided into nine groups: A (a), B, C, D, E, b, c, d and e. The A (a) group was the control. In the B, C, D and E groups (continuing impact group), PSCs were treated with four concentrations of TGF-β1 (0.5, 1, 5, and 10 ng/ml). In the b, c, d and e groups (transient impact group), TGF-β1 was added to the culture medium 30 minutes before the total RNA was extracted, to make the concentrations of TGF-β1 in the culture medium 0.5, 1, 5, and 10 ng/ml.
Immunostaining
Cells grown on coverslips were washed and fixed in 10% neutral medium. Endogenous peroxide was blocked with 3% H2O2. Nonspecific proteins were blocked with (bovine serum albumin) BSA. Immunostaining was performed using anti-rat desmin and α-SMA monoclonal antibodies with the ultrasensitive S-P kit according to the manufacturer's instructions.
Reverse transcription-polymerase chain reaction (RT-PCR)
Smad3 and Smad7 mRNA levels were detected by RT-PCR. Total RNA was extracted using Trizol reagent according to the manufacturer's instructions. cDNA was synthesized from 2 μg total RNA followed by PCR using an RT-PCR kit according to the manufacturer's instructions. All primers are listed in Table. The PCR conditions were as follows: 94 ℃ for 2 minutes for predenaturing, then 38 cycles for denaturing at 94 ℃for 1 minute, annealing at 57 ℃ for 30 seconds, and polymerization at 72 ℃ for 1 minute. PCR products were separated by electrophoresis on a 2% agarose gel followed by ethidium bromide staining. The target bands were analyzed densitometrically using a GS-800 calibrated densitometer and gel analyzing software (Bio-Rad, CA), and the results were calculated as the ratio of the OD value relative to β-actin.
Western blotting analysis
Smad3 and Smad7 protein levels were detected by Western blotting analysis. Aliquots of cell lysates containing 50 μg of proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with a Tris-buffered saline Tween-20 buffer (10 mmol/L Tris-HCl, 100 mmol/L NaCl, 0.1% Tween-20) containing 5% skimmed milk, incubated with antibodies against p-Smad3, Smad3, andSmad7 (1∶200 dilution) overnight, and followed by the addition of HRP-linked IgG (1∶4000 dilution) and ECL visualization of the bands.

Table. Sequences of Smad3, Smad7 and β-actin primers
Statistical analysis
All data were expressed as mean±SD and were analyzed with the statistical software SPSS 10.0 for Windows. One-way analysis of variance was used to determine the statistically significant differences among groups, and the averages for two different groups were analyzed with Student'sttest. AP<0.05 was considered statistically significant.
Results
Histological appearance
The freshly isolated PSCs appeared small and round on light refraction microscopy. Trypan blue staining showed that the vitality was more than 95%. Cells began to adhere within 1 hour after seeding and extended to typical stellate cells in 24 hours (Fig. 1A). The cell volume was clearly increased in completely activated PSCs (Fig. 1B). Desmin staining showed brown-yellow fibers distributed in the cytoplasm of PSCs (Fig. 1C), and α-SMA staining showed that α-SMA expression appeared in the activation process (Fig. 1D).
Changes in Smad3 and Smad7 mRNA levels
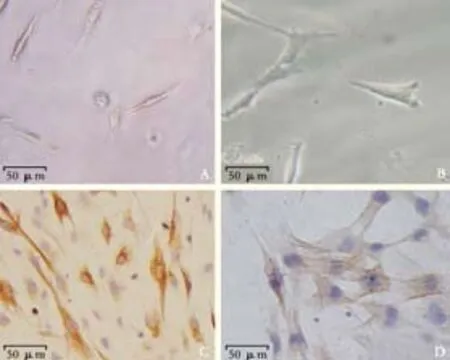
Fig. 1. Histological appearance of PSCs. A: freshly isolated PSCs (original magnification ×200); B: activated PSCs (original magnification ×400); C: Desmin immunostaining (original magnification × 200); D: α-SMA immunostaining (original magnification ×400).
Half an hour after PSC isolation, Smad3 mRNA was not detected under normal culture conditions, but Smad7 mRNA was highly expressed (group a) (Fig. 2). However, Smad3 mRNA was expressed in small quantities in cells treated with various concentrations of TGF-β1 (groups c, d, e). The expression of Smad7 mRNA decreased gradually along with the increasing concentration of TGF-β1, and expression was the lowest when the concentration of TGF-β1 reached 10 ng/ml. Seven days after PSC isolation, Smad3 mRNA was spontaneously expressed in untreated cells; however, the expression was inhibited in cells treated with 0.5 ng/ml TGF-β1 and enhanced in those treated with 10 ng/ml (Fig. 3). The expression of Smad7 mRNA decreased gradually with the increasing concentration of TGF-β1. In groups A,B, C, D and E, we further analyzed the Smad3 mRNA expression in different TGF-β1 concentrations at different time points (Fig. 4). Smad3 mRNA expression was observed in both the TGF-β1-treated and untreated groups after 96 hours (P<0.05). After 7 days, however, Smad3 mRNA expression was inhibited in the group treated with high-dose TGF-β1 (P<0.05). These findings suggest that TGF-β1 promotes the expression of Smad3 and inhibits the expression of Smad7 during the activation of PSCs. However, high-dose TGF-β1 inhibits Smad3 mRNA expression.
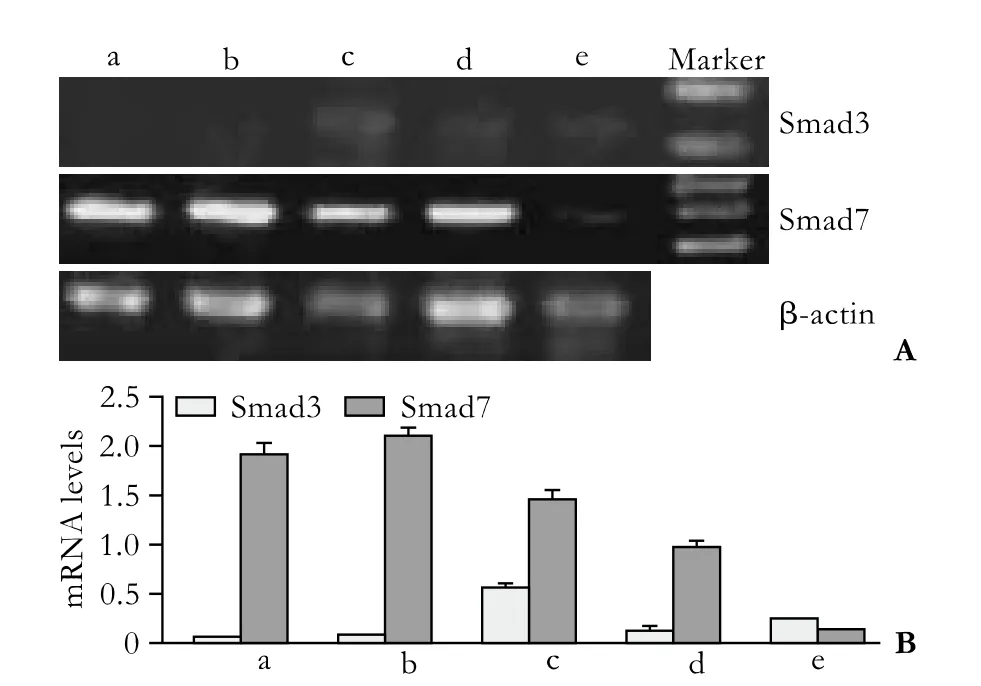
Fig. 2. Transient impact group: Smad3 and Smad7 mRNA expression 0.5 hours after PSC isolation. a: normal control; b-e: TGF-β1 was added to the culture medium 30 minutes before the total RNA was extracted, to make the concentrations of TGF-β1 0.5, 1, 5 and 10 ng/ml.
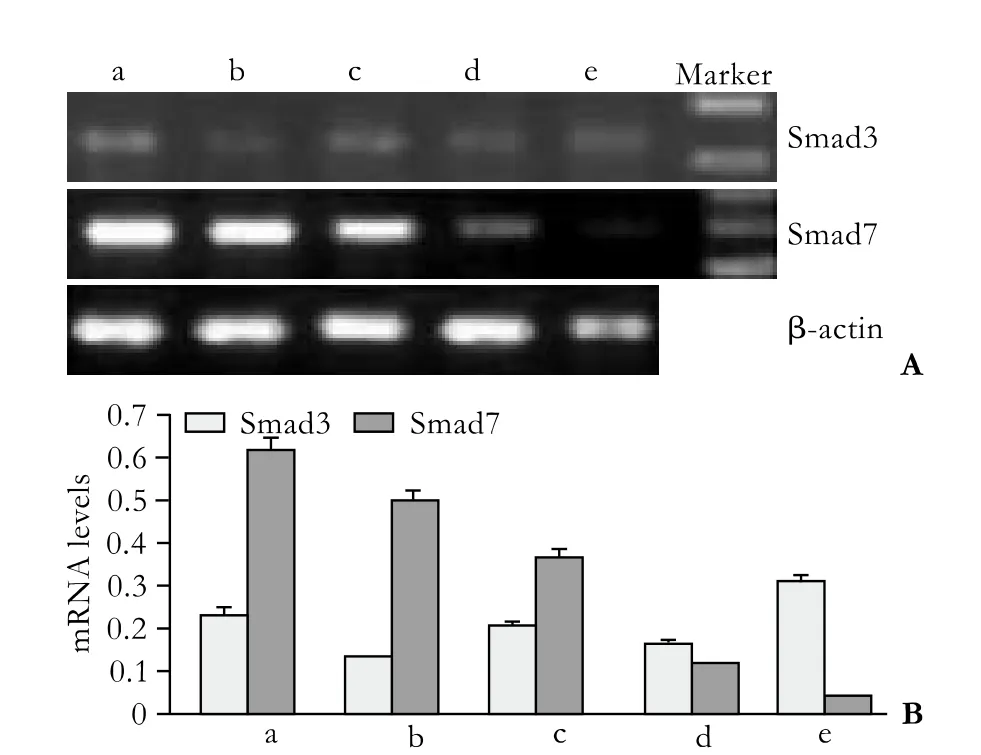
Fig. 3. Transient impact group: Smad3 and Smad7 mRNA expression 7 days after PSC isolation. a: normal control; b-e: TGF-β1 was added to the culture medium 30 minutes before the total RNA was extracted, to make the concentrations of TGF-β1 0.5, 1, 5 and 10 ng/ml.
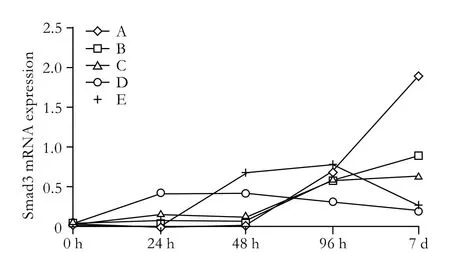
Fig. 4. Continuing impact group: Smad3 mRNA expression at different time points in different TGF-β1 concentrations. A: normal control; B-E: PSCs were cultured in medium supplemented with TGF-β1 (0.5, 1, 5 and 10 ng/ml).
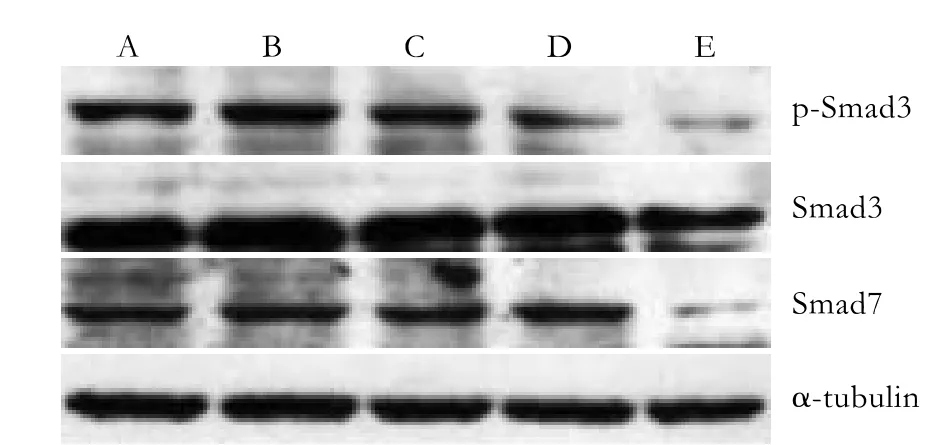
Fig. 5. Continuing impact group: p-Smad3, Smad3, and Smad7 protein expression 7 days after PSC isolation. A: normal control; B-E: PSCs were cultured in medium supplemented with TGF-β1 (0.5, 1, 5 and 10 ng/ml).
Changes in p-Smad3, Smad3 and Smad7 protein levels
Western blotting analysis showed an excessive expression of Smad7 in the quiescent stage of PSCs but no Smad3 expression. With increasing activation of the cells, the Smad3 protein began to be expressed. In cells treated with 5 ng/ml TGF-β1 (group D), Smad3 was expressed in small quantities after 24 hours, and its expression increased continuously; however, the expression levels did not significantly differ among the groups. The expression of Smad3 in cells treated with 10 ng/ml TGF-β1 (group E) after 7 days was significantly less than that in the other groups. Moreover, p-Smad3 protein was inhibited distinctly (Fig. 5,P<0.05). The Smad7 expression decreased along with the activation of PSCs. Compared with the other groups, the expression of Smad7 in group E was significantly inhibited 7 days after isolation; its expression in the other groups showed no evident differences.
Discussion
An isolated PSC in the process of culture can be activated automatically to transform into a stellatelike cell similar to a myofibroblast.[3]It is generally considered that TGF-β1 is the primary biological factor that promotes the biosynthesis of ECM such as collagen and the formation of pancreatic fibrosis by activating PSCs.[12]In addition, TGF-β1 can promote the synthesis of collagen, reduce the expression of matrix metalloproteinase (MMP)-3 and MMP-9, and inhibit cell proliferation.[13]Only by the effect of corresponding receptors on the membrane and the downstream effect of molecules can the activation signal of PSCs be produced by TGF-β1.
The Smad protein is the key substrate for the TGF-β1 kinase receptor family,[14]and the structure, composition, and basic functions of Smad have been extensively studied. The 8 members of the Smad family which have been discovered can be classified into 3 categories as follows: 1) R-Smad (Smad1, 2, 3, 5, 8) activated by receptor kinase, which is the direct substrate of the TGF-β1 kinase receptor family; 2) Co-Smad (Smad4) which can connect with R-Smad and participate in signal transduction, but Smad4 does not mediate signal transduction; and 3) I-Smad (Smad6, 7) which can activate R-Smad by blocking the receptor so as to inhibit the signal transduction of the other categories. Smad3 and Smad7 included in our study belong to R-Smad and I-Smad, respectively.
Smad3 is the key factor in the TGF-β1/Smad3 signal pathway, the expression level of which directly affects the activation and expression of collagen. Moreover, Smad3 expression can also be regulated by TGF-β1. In early PSCs in the course of activation, TGF-β1 promoted Smad3 to express ahead of schedule. Moreover, Smad3 was expressed first when the concentration of TGF-β1 was 5 ng/ml. When the concentration increased, the Smad3 expression decreased because of the negative feedback regulation of the TGF-β1/Smad3 signal pathway; however, the specific transduction pathway has not been clarified so far.[15,16]In the early activation process of PSCs, TGF-β1 promotes Smad3 expression and thus accelerates collagen synthesis.
Uemura et al[17]found that excessive expression of the Smad3 protein activates PSCs and produces more collagens and fibrins, which is similar to our results. Other researchers selected Smad3-deficient mice in order to study the TGF-β1/Smad3 transduction pathway and found that the transduction pathway is inhibited so as not to lead to pulmonary fibrosis.[18-20]It has been suggested that the TGF-β1/Smad3 transduction pathway plays a key role in collagen synthesis. After PSCs are activated completely, they secrete TGF-β1.[6]Moreover, because the concentration of TGF-β1 in the culture medium increased distinctly, Smad3 expression could be inhibited by means of the negative feedback regulation of the TGF-β1/Smad3 signal pathway.
During the entire process of activation, the PSCs activated by TGF-β1 inhibited Smad7 expression. Smad7 belonged to the I-Smad group and could compete with the members of the R-Smad group for receptors so as to block TGF-β1/Smad3 signal transduction.[21]The PSCs isolated in the quiescent stage demonstrated an excessive expression of Smad7. In the normal tissue of the pancreas, excessive Smad7 expression keeps PSCs in the quiescent stage. With the activation of PSCs, the expression of Smad7 decreased and that of Smad3 increased gradually; thus, the inhibition caused by Smad7 was weakened. Meanwhile, the TGF-β1/Smad3 transduction pathway transmits signals to promote collagen synthesis and cell activation. Because of the negative feedback regulation of the TGF-β1/Smad3 signal pathway, the expression imbalance between Smad3 and Smad7 could be improved when PSCs were activated completely. The expression of Smad7 increased with PSC activation; in addition, the PSCs secreted TGF-β1 in order to promote the formation of ECM, which affected the expression of Smad3 and Smad7.
Gene transfection technology can be used to obtain a low expression of Smad3 or an excessive expression of Smad7 to continuously maintain PSCs in the quiescent stage or to transform them from the activating stage to the quiescent stage, thus inhibiting the TGF-β1/ Smad3 transduction pathway in order to reduce ECM synthesis. In liver diseases, high expression of Smad3 in hepatic stellate cells can lead to hepatocirrhosis. PSCs whose function is similar to that of hepatic stellate cells also play an important role in pancreatic fibrosis. The TGF-β1/Smad3 transduction pathway is imperative for PSCs to synthesize fibrous tissue; further, the low expression of Smad3 and the excessive expression of Smad7 can clearly inhibit fiber synthesis so as to inhibit pancreatic fibrosis. Bonniaud et al[19]used TGF-β1 to induce the formation of pulmonary fibrosis in Smad3-deficient mice and found that the transduction pathway and collagen synthesis are inhibited so as not to lead to pulmonary fibrosis. Nakao et al[22]selected mice that expressed Smad7 excessively, induced pulmonary fibrosis in them with bleomycin, and found that the expression and activity of Smad2 and Smad3 were inhibited and fibrin synthesis was reduced. Lan et al[23]used transgenic technology to transfect rat models with obstructive nephropathy so as to increase the Smad7 expression in rats and found that the degree of renal fibrosis was significantly less than that of the control group. All of these results suggest that low expression of Smad3 or excessive expression of Smad7 inhibits the TGF-β1/Smad3 transduction pathway; therefore, it may be possible to control and treat chronic pancreatitis.
In conclusion, the two signal molecules Smad3 and Smad7, which are antagonistic, play important roles in the activation of PSCs. An expression imbalance between them promoted the activation of PSCs. The gradual expression of Smad3 promoted cell activation, and Smad7 was inhibited in the course of cell activation; Smad3 expression was promoted and Smad7 expression was inhibited by TGF-β1 in the course of PSC activation. Smad3 expression might be inhibited by TGF-β1 at high concentrations by means of the negative feedback regulation of the TGF-β1/Smad3 transduction pathway; however, further studies are required to elucidate the details of this regulation.
Funding:This study was supported by grants from the Natural Science Foundation of Jiangsu Province, China (No. BK2006241) and the Foundation for Talents in Six Fields of Jiangsu Province (No. 07-B-038).
Ethical approval:The Animal Care Committee of Nanjing Medical University approved this study.
Contributors:QZY proposed the study. ZZW wrote the first draft. PQ and ZZW analyzed the data. All authors contributed to the design and interpretation of the study and to further drafts. MY is the guarantor.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Watari N, Hotta Y, Mabuchi Y. Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn 1982;58: 837-858.
2 Ikejiri N. The vitamin A-storing cells in the human and rat pancreas. Kurume Med J 1990;37:67-81.
3 Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998;115:421-432.
4 Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43: 128-133.
5 Shinji T, Ujike K, Ochi K, Kusano N, Kikui T, Matsumura N, et al. Establishment of a novel collagenase perfusion method to isolate rat pancreatic stellate cells and investigation of their gene expression of TGF-beta1, typeicollagen, and CTGF in primary culture or freshly isolated cells. Acta Med Okayama 2002;56:211-218.
6 Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest 2007;117:50-59.
7 Apte MV, Pirola RC, Wilson JS. Battle-scarred pancreas: role of alcohol and pancreatic stellate cells in pancreatic fibrosis. J Gastroenterol Hepatol 2006;21:S97-101.
8 Phillips PA, McCarroll JA, Park S, Wu MJ, Pirola R, Korsten M, et al. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut 2003;52:275-282.
9 Apte MV, Wilson JS. Mechanisms of pancreatic fibrosis. Dig Dis 2004;22:273-279.
10 Bachem MG, Zhou Z, Zhou S, Siech M. Role of stellate cells in pancreatic fibrogenesis associated with acute and chronic pancreatitis. J Gastroenterol Hepatol 2006;21:S92-96.
11 Zhou LA, Yang GY, Qian ZY. Isolation of pancreatic stellate cells using collagenase perfusion method through abdominal aorta cannulation and its culture and characterization. Nanjing Yike Daxue Xuebao 2007;27:11-14.
12 Shimosegawa T, Kume K, Satoh K. Chronic pancreatitis and pancreatic cancer: prediction and mechanism. Clin Gastroenterol Hepatol 2009;7:S23-28.
13 Aoki H, Ohnishi H, Hama K, Ishijima T, Satoh Y, Hanatsuka K, et al. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol 2006;290: C1100-1108.
14 Stopa M, Benes V, Ansorge W, Gressner AM, Dooley S. Genomic locus and promoter region of rat Smad7, an important antagonist of TGFbeta signaling. Mamm Genome 2000;11:169-176.
15 Watanabe Y, Itoh S, Goto T, Ohnishi E, Inamitsu M, Itoh F, et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol Cell 2010;37:123-134.
16 Ehnert S, Knobeloch D, Blankenstein A, Müller A, Böcker U, Gillen S, et al. Neohepatocytes from alcoholics and controls express hepatocyte markers and display reduced fibrogenic TGF-beta/Smad3 signaling: advantage for cell transplantation? Alcohol Clin Exp Res 2010;34:708-718.
17 Uemura M, Swenson ES, Gaça MD, Giordano FJ, Reiss M, Wells RG. Smad2 and Smad3 play different roles in rat hepatic stellate cell function and alpha-smooth muscle actin organization. Mol Biol Cell 2005;16:4214-4224.
18 Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc Am Thorac Soc 2006;3:696-702.
19 Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampf liM, et al. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol 2004;173:2099-2108.
20 Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 2002;282:L585-593.
21 Hama K, Ohnishi H, Aoki H, Kita H, Yamamoto H, Osawa H, et al. Angiotensin II promotes the proliferation of activated pancreatic stellate cells by Smad7 induction through a protein kinase C pathway. Biochem Biophys Res Commun 2006;340:742-750.
22 Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, et al. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest 1999;104:5-11.
23 Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ, et al. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol 2003;14:1535-1648.
January 28, 2010
Accepted after revision June 27, 2010
Author Affiliations: Department of General Surgery, First Affiliated Hospital, Nanjing Medical University, Nanjing 210029, China (Qian ZY, Peng Q, Zhang ZW, Zhou LA and Miao Y)
Zhu-Yin Qian, PhD, Department of General Surgery, First Affiliated Hospital, Nanjing Medical University, Nanjing 210029, China (Tel: 86-25-83718836; Fax: 86-25-83724440; Email: qianzhusilver@163. com)
© 2010, Hepatobiliary Pancreat Dis Int. All rights reserved.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- News
- Advances in prognostic factors in acute pancreatitis: a mini-review
- Hepatocellular carcinoma metastatic to the kidney mimicking renal oncocytoma
- Simultaneous breast and ovarian metastasis from gallbladder carcinoma
- An eight-year journey of Hepatobiliary & Pancreatic Diseases International
- Interferon and lamivudine combination therapy versus lamivudine monotherapy for hepatitis B e antigen-negative hepatitis B treatment: a meta-analysis of randomized controlled trials