HPLC法同时测定饲料预混料中维生素C(VC)和VC磷酸酯的含量
2010-04-28湖南农业大学食品科技学院张春艳
湖南农业大学食品科技学院 张春艳
广东海大集团畜牧水产研究中心 周孝治*
维生素C(VC)又称L-抗坏血酸,是动物生长发育过程中不可缺少的营养素之一(张辉等,2009),其结构中存在易氧化基团,在饲料加工、储藏和使用过程中稳定性较差(葛建东和杨凌,1998)。实际应用中,常将VC酯化为稳定的VC磷酸酯,但由于其酯化程度及加工工艺不同,饲料添加剂或预混料中VC磷酸酯常为单体VC和VC磷酸酯的混合物,因而其稳定性或利用率存在差异(张玮和尚平,2005)。
目前饲料添加剂中VC检测主要采用氧化还原滴定法和邻苯二胺荧光法,VC磷酸酯检测主要采用酶解法和紫外分光光度法。由于饲料预混料中干扰物质较多,采用这些方法操作步骤复杂,且不能区分单体VC和VC磷酸酯的含量,因而对不同厂家生产的饲料预混料中VC含量容易产生误判(张丽萍和吴小春,2002)。本文旨在建立饲料预混料中VC和VC磷酸酯含量同时测定的高效液相色谱(HPLC)法,以期获得简单快速、干扰少、回收率高、检测结果准确可靠的方法。
1 材料与方法
1.1 仪器与试剂 Agilent1200型高效液相色谱仪,配有二极管阵列检测器(DAD),Agilent Chem-Station色谱工作站;HERMLE-Z323型高速离心机;Sartorius-CP225D型电子分析天平;SCQ-3201A超声波清洗仪。
VC标准品(Dr.Ehrenstorfer公司,纯度 ≥99.0%),VC磷酸酯钠盐标准品(Fluka公司,纯度≥ 98.0%),乙腈为色谱纯,磷酸二氢钾、磷酸为分析纯,实验用水为Millipore超纯水。
1.2 溶液配制 磷酸缓冲液的配制:准确称取40.83 g磷酸二氢钾用超纯水溶解稀释至1000 mL,用85%磷酸调节pH至4.0,然后用0.45 μm滤膜过滤,配制成0.3 mol/L磷酸二氢钾(pH 4.0)的缓冲溶液。
混合标准储备液的配制:准确称取VC和VC磷酸酯钠盐标准品各100.00 mg于100 mL棕色容量瓶中,用磷酸缓冲液溶解并定容至刻度,即成1000.0 mg/L的VC和VC磷酸酯钠盐混合标准储备液,于4℃冰箱避光保存。
1.3 试验样品 饲料预混料,由广东某研究中心提供。
1.4 试验方法
1.4.1 色谱分析条件 色谱柱:Lichrospher-NH2分析柱(250 mm×4.0 mm i.d.,5 μm);流动相:乙腈-磷酸缓冲液(30∶70,V/V);检测器:DAD 检测器,检测波长为 254 nm;柱温:25℃;进样量:10 μL;流速:1.0 mL/min。
1.4.2 样品的预处理 称取样品0.5~1 g(精确至0.0001 g)于100 mL棕色具塞三角瓶中,加入流动相50 mL,摇匀,于25℃水浴超声提取15 min,冷却静置2 min,取上层提取液约30 mL转入离心管中,以4000 r/min离心10 min,上清液用0.22 μm微孔滤器过滤,进HPLC分析。
1.4.3 标准曲线的绘制 用流动相将VC和VC磷酸酯钠盐混合标准储备液逐级稀释得到浓度为5.0、10.0、25.0、50.0、100.0 mg/L 和 200.0 mg/L 的一系列标准工作液,在选定的色谱条件下,将浓度由低到高进样检测,以峰面积-浓度作图,得到标准曲线回归方程。
1.4.4 样品的测定 按外标法,将各试样经HPLC检测所得的VC和VC磷酸酯钠盐色谱峰面积分别代入相应的标准曲线方程,得到样品中VC和VC磷酸酯钠盐的含量,其中VC磷酸酯钠盐含量再乘以VC磷酸酯钠盐转换为VC磷酸酯的系数0.7951,求得样品中VC磷酸酯的含量。
2 结果与讨论
2.1 样品预处理条件的选择
2.1.1 提取液的选择 由于VC和VC磷酸酯属于极性化合物,其中VC溶液热稳定性较差,在中性或碱性水溶液中易被氧化,但在酸性溶液中比较稳定,实验过程中分别用磷酸缓冲液(pH 4.0)和流动相作为提取液,考察了提取液对试样提取的影响。结果显示,采用流动相作为试样提取液时提取效果良好,且色谱峰形尖锐对称,因此选用流动相作为试样提取液。
2.1.2 提取条件的选择 实验选用流动相作为试样提取液,对提取温度(25、35、45、50 ℃)和提取时间(5、10、15、20 min) 两个条件参数进行了优化。结果表明:随着温度的升高,VC磷酸酯的提取效率加快,但单体VC的稳定性减弱;随着提取时间的增加,试样提取效率有所增加,当提取时间超过15 min时,提取液中杂质干扰增多,最终确定参数为:提取温度25℃,提取时间15 min。
2.2 色谱条件的选择
2.2.1 检测波长的选择 采用流动相作为背景缓冲液,对VC和VC磷酸酯标准液和样品溶液用二极管阵列检测器在190~400 nm进行全波长紫外扫描。结果表明,VC和VC磷酸酯两种物质在254 nm处相对吸收最大,且杂峰干扰少,本实验选择检测波长为254 nm。
2.2.2 色谱柱的选择 考察了 C18、C8和 NH2分析色谱柱对VC和VC磷酸酯分离效果的影响,由于VC和VC磷酸酯极性较强,在C18和C8分析柱上保留时间短,不能很好的与杂峰分离,但在具有极性官能团的NH2色谱柱上,VC、VC磷酸酯与杂峰之间分离效果良好,相对保留时间较长,故选择NH2色谱柱作为分离柱。
2.2.3 流动相的选择 由于VC在酸性介质中比较稳定,实验过程中考察了流动相磷酸缓冲液不同 pH 值(3.0、4.0、5.0)对色谱分离效果的影响。结果表明pH为4.0时,VC和VC磷酸酯吸收值较大,灵敏度高,色谱峰形对称,因此缓冲液pH选择为4.0。
实验过程中还分别通过调节磷酸缓冲液不同浓度(0.1、0.2、0.3 mol/L)和乙腈-磷酸缓冲液不同比例(20∶80、30∶70、40∶60,V/V)来改善色谱峰的分离效果。结果显示,随着磷酸缓冲液浓度的升高,色谱峰形和分离度越好;随着乙腈比例的升高,保留时间缩短,杂质干扰较大。当乙腈-磷酸缓冲液(0.3 mol/L)比例为 30∶70(V/V)时,目标峰与样品中的杂峰分离良好,色谱峰形尖锐、对称(见图1),在此色谱条件下VC和VC磷酸酯的保留时间分别为(4.10±0.05)min 和(8.12±0.05)min。
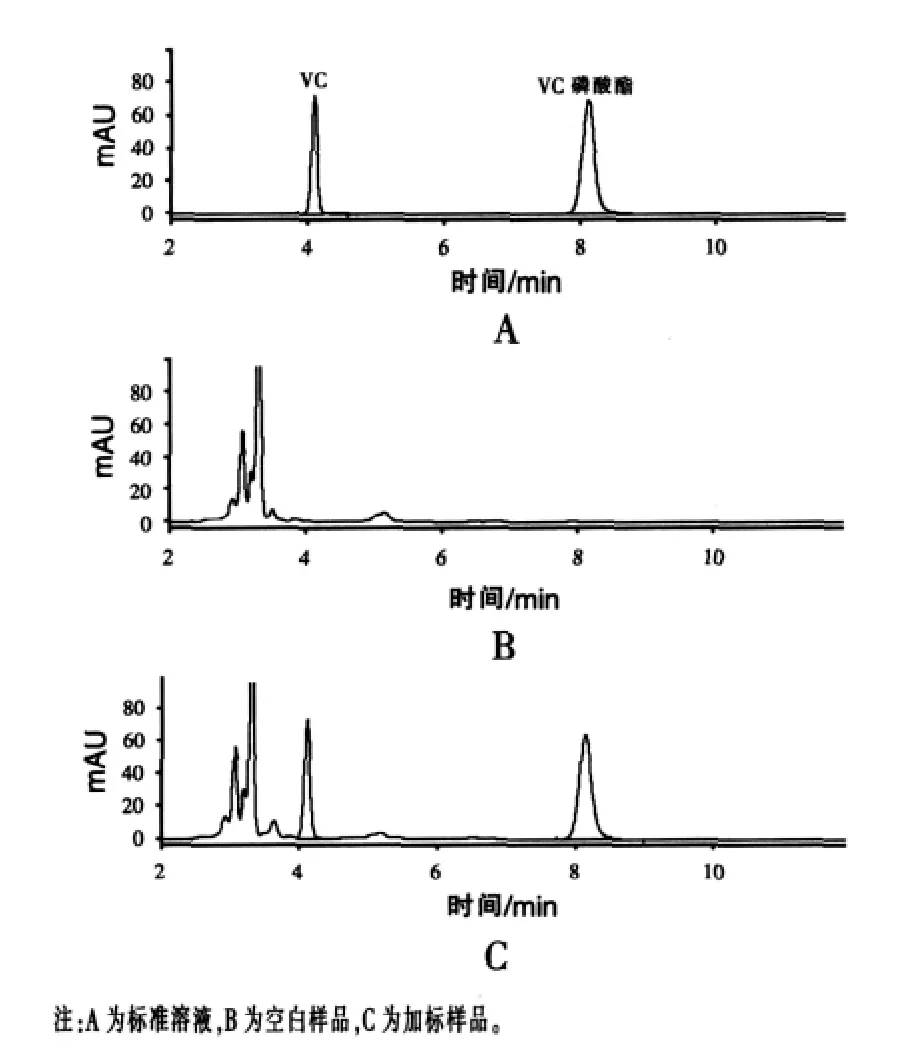
图1 不同样品中VC和VC磷酸酯的高效液相色谱图
2.3 标准曲线和方法检出限 在上述色谱条件下, 对 5.0、10.0、25.0、50.0、100.0 mg/L 和 200.0 mg/L的VC和VC磷酸酯混合标准工作液进样分析,以HPLC测得的峰面积为纵坐标(y),相应的浓度为横坐标(x)绘制标准曲线,得出VC和VC磷酸酯的线性回归方程、相关系数和线性范围,根据3倍信噪比(S/N=3)可计算出方法的检出限,结果见表1。试验结果表明,浓度为5.0~200.0 mg/L时,VC和VC磷酸酯线性关系良好,检出限分别为0.05 mg/L和0.1 mg/L。
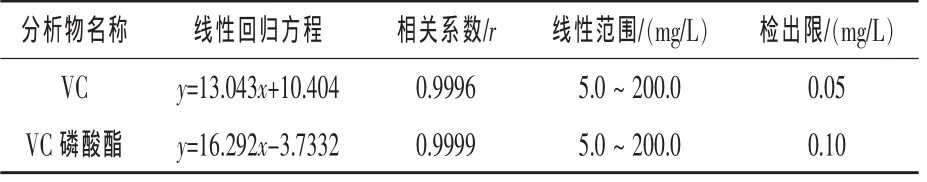
表1 VC和VC磷酸酯的线性回归方程及检出限
2.4 方法的准确度和精密度 对未添加VC和VC磷酸酯的空白饲料预混料进行加标样品回收率和精密度实验,选择了 100.0、1000.0、5000.0 mg/kg三个添加水平,每个浓度平行测定5次,重复3 d,结果见表2。
从表2中的数据可以看出,添加浓度为100.0~5000.0 mg/kg时,饲料预混料样品中VC和VC磷酸酯的平均回收率为95.6%~99.1%,日内相对标准偏差为0.97%~1.76%,日间相对标准偏差为1.98%~3.84%,方法检测的准确度和精密度良好,满足分析的要求。
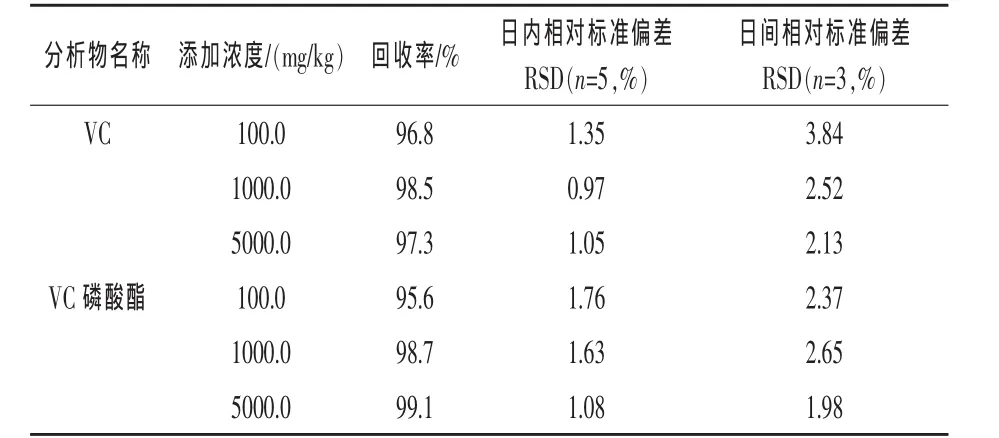
表2 饲料预混料中VC和VC磷酸酯的回收率和精密度测定
2.5 样品测定 按本文所建立的方法测定了A、B、C、D、E五种不同饲料预混料样品中VC和VC磷酸酯的含量,将VC磷酸酯含量转换为VC含量(VC磷酸酯转换为VC的系数按0.6876计),与所测单体VC含量之和得到总VC含量。结果如表3所示,五种不同饲料预混料添加的总VC含量大致相同,约为5500 mg/kg,但其中单体VC含量和VC磷酸酯含量却各不相同,存在较大差异。

表3 不同饲料预混料中VC、VC磷酸酯和总VC含量
3 结论
采用高效液相色谱法检测饲料预混料中VC和VC磷酸酯含量,其检测结果准确度高,平均回收率在95.6%~99.1%,重现性好,日内相对标准偏差在0.97%~1.76%,日间相对标准偏差在1.98%~3.84%,样品的预处理简单快速,不受样品中其他组分的干扰,适用于饲料预混料中VC和VC磷酸酯的快速定量分析测定。
[1]葛建东,杨凌.饲料工艺对预混料中维生素的影响[J].饲料研究,1998,1:15~17.
[2]张辉,牟振波,单安山,等.鱼类的维生素 C 营养[J].饲料工业,2009,30(8):48~50.
[3]张丽萍,吴小春.国内维生素C仪器定量分析进展[J].四川轻化工学院学报,2002,12(15):34 ~42.
[4]张玮,尚平.稳定性维生素C源的提纯工艺研究[J].石家庄职业技术学院学报,2005,6:24 ~25.
