自固化磷酸钙微球的制备与表征*
2010-03-16叶建东李江波周子强于涛
叶建东 李江波 周子强 于涛
(1.华南理工大学材料科学与工程学院,广东广州 510640;2.广州市红十字会医院骨科,广东广州 510270)
由于具有较好的流动性、较高的堆积密度、较好的填充性和非致炎症性[1],表面光洁且球形度好的羟基磷灰石(HA)微球颗粒骨缺损填充材料受到了广泛关注.同时,HA微球具有骨传导能力[2],有助于骨组织的长入[3],因此,HA微球在临床口腔手术中得到了应用,如牙根管、拔牙窝充填、牙槽骨扩充以及牙周修复等[4-5].然而,HA微球在制备过程中若不经烧结处理,在生理环境下很快便会发生崩解,导致体系失效[6];若经烧结处理,一方面微球成为结晶完善的六方晶格HA的较致密多晶体,具有较高的稳定性,其在体内的降解和吸收变得极为缓慢,但另一方面又难以在微球制备过程中加入功能性组分(如载药).自固化磷酸钙(磷酸钙骨水泥,CPC)具有可任意塑形性,利用其自固化特性,不仅可以很方便地在体温下或室温下根据临床需要制成不同大小、不同形状的修复体,而且可在制备过程中添加功能性成分.
自固化磷酸钙是由一种或多种磷酸盐粉末与液相 (蒸馏水、溶液或血液等)组成的新型骨修复材料.固液两相混合后可在人体生理条件下 (37℃)发生水化反应而凝固,最终产物为骨骼的主要无机成分——羟基磷灰石[7].HA具有良好的生物相容性、可塑形性、可降解性和骨传导性,可用于修复骨质缺损和促进骨折愈合,是临床骨组织缺损修复的理想材料之一.
文中采用反相乳液法,利用磷酸钙骨水泥的自固化特性,制备了粒径约为 100~1000μm的自固化磷酸钙微球,并对微球的结构与性能进行测试和表征,为拓宽磷酸钙骨水泥的应用范围提供依据.
1 材料与方法
1.1 原料
自固化磷酸钙为笔者所在课题组自行研发的PCCP-DCPA体系骨水泥,由PCCP(部分结晶磷酸钙)和DCPA(无水磷酸氢钙,上海试四赫维化工有限公司产品,分析纯)按质量比1∶1均匀混合得到,其中PCCP是通过将钙盐溶液和磷酸盐溶液反应形成的沉淀进行冷冻干燥和低温煅烧等后期处理得到[8].其他主要原料如下:明胶,国药集团化学试剂有限公司产品,化学纯;变性淀粉,华南理工大学轻工与食品学院提供,食品级;大豆油,中粮集团产品,食品级;丙酮,广州市东红化工厂产品,分析纯;无水乙醇,天津市富宇精细化工有限公司产品,分析纯.
1.2 自固化磷酸钙微球的制备
自固化磷酸钙微球通过反相乳液法制备,具体方法是将液相成分(明胶/变性淀粉溶液)和固相颗粒(CPC粉末)按液固比2.0∶1~3.0∶1(液相体积以mL计,固相质量以g计)充分混合,制得自固化磷酸钙浆体,再将浆体搅拌分散于 4℃的大豆油中(搅拌速度150~300 r/m in),30min后,迅速用300目滤布滤除大豆油,初步分离出微球,并用丙酮和无水乙醇反复洗微球数次,随再将得到的微球在温度为37℃、湿度为 97%的恒温恒湿箱中养护水化48h,干燥、收集保存微球.其中,液相成分的制备方法为:将明胶颗粒加入至45℃的去离子水中溶胀30m in(明胶质量浓度为3.0~8.0g/mL),待明胶颗粒完全溶解后,往溶液中加入变性淀粉(变性淀粉的质量浓度为5g/L),充分溶解得到明胶/变性淀粉溶液.
固定其他因素,选取对微球粒径影响较为显著的搅拌速度(A,r/min)、明胶质量浓度(B,g/m L)、液固比(C)和油水相体积比(D),来调控微球的粒径.采用正交试验表L9(34)来安排试验,以微球的粒径和相对跨距(SPAN)作为考察对象,按正交试验结果得出不同粒径微球的制备条件.正交试验因素水平见表1.
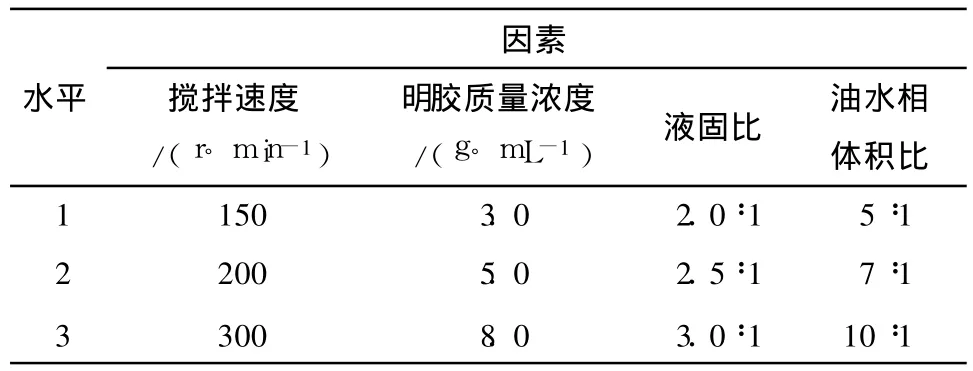
表1 正交试验因素水平表Table 1 Factors and levels of orthogonal test
1.3 微球的表征
用激光粒度仪(Master Sizer 2000,英国Malven Instruments公司)测定微球粒径和粒度分布.中位径D50表征微球粒度,相对跨距表征粒度分布,其中 D90、D10、D50分别为有 90%、10%和50%的粒子的粒径小于该值的粒径.
用扫描电子显微镜(Quanta 200,荷兰FEI公司)观察微球的表面形貌和断面形貌.
用X射线衍射仪(X'Pert Pro,荷兰Panlytical公司)对由微球制备的粉末进行物相分析,Cu靶Kα射线,电压40 kV,电流4mA.通过FT-IR(Avatar 360,美国Nicolet公司)辅助分析微球的结构.
用Poremaster 33/60型压汞仪(美国Quantachrome公司)测定微球的孔隙率和孔径分布.
采用固定漏斗法测量微球的休止角 θ′,计算公式为tanθ′=2H/D,H和D分别代表微球自由堆积形成的堆的高度和直径.
1.4 微球抗崩解性及力学性能的测试
以变性淀粉为抗崩解剂,分别对不含变性淀粉的CPC微球、含变性淀粉的CPC微球、含变性淀粉的HA微球进行抗崩解性定性测试(3组微球分别记为a、b、c).测试方法为:将a、b、c 3组样品浸泡在纯水中,同时置于摇床中,在37℃下以120r/min的速度振荡,分别于0、3、6、12h时取出拍照.c组HA微球是以HA粉末[10]为原料,HA微球的制备方法和实验中自固化磷酸钙微球的制备方法完全相同.
用5567型万能试验机(美国Instron公司)测定微球的力学性能,设计φ=10mm、h=20mm的推杆式圆柱体不锈钢模具,在空腔中填满微球,垂直方向上的推杆与试验机的抗压传感器接触待测.
1.5 微球的细胞毒性实验
在DME(Dulbecco's Modified Eagle)培养基中加入小苏打、葡萄糖、胎牛血清、青霉素和链霉素,配制小苏打质量浓度为1.5mg/mL、葡萄糖质量浓度为4.5mg/m L、胎牛血清浓度(体积分数)为10%、青霉素浓度为100units/mL、链霉素质量浓度为100μg/mL的培养液,用来培养新西兰大白兔的骨髓间充质干细胞.微球用70%(体积分数)的乙醇消毒后,再用PBS(磷酸盐缓冲溶液)冲洗,随后在培养基中浸泡12 h备用.
微球的细胞毒性通过MTT(3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐)法测得[11].具体步骤为:将微球置于24孔细胞培养板中,按浓度1×104个/mL在微球上植入细胞,将培养板置于 37℃、湿度95%的恒温恒湿箱中培养一周,其中CPC微球组为含有添加剂的自固化磷酸钙微球,CPC对照组为纯自固化磷酸钙球状固化体.用酶标仪(3001型,美国Thermo Fisher公司)在490nm波长处测定其吸光度(光密度),吸光度的高低间接反映活细胞的数量及活性.
2 结果与讨论
2.1 正交试验结果与分析
不同工艺参数下制备的微球的中位径和相对跨距如表2所示.
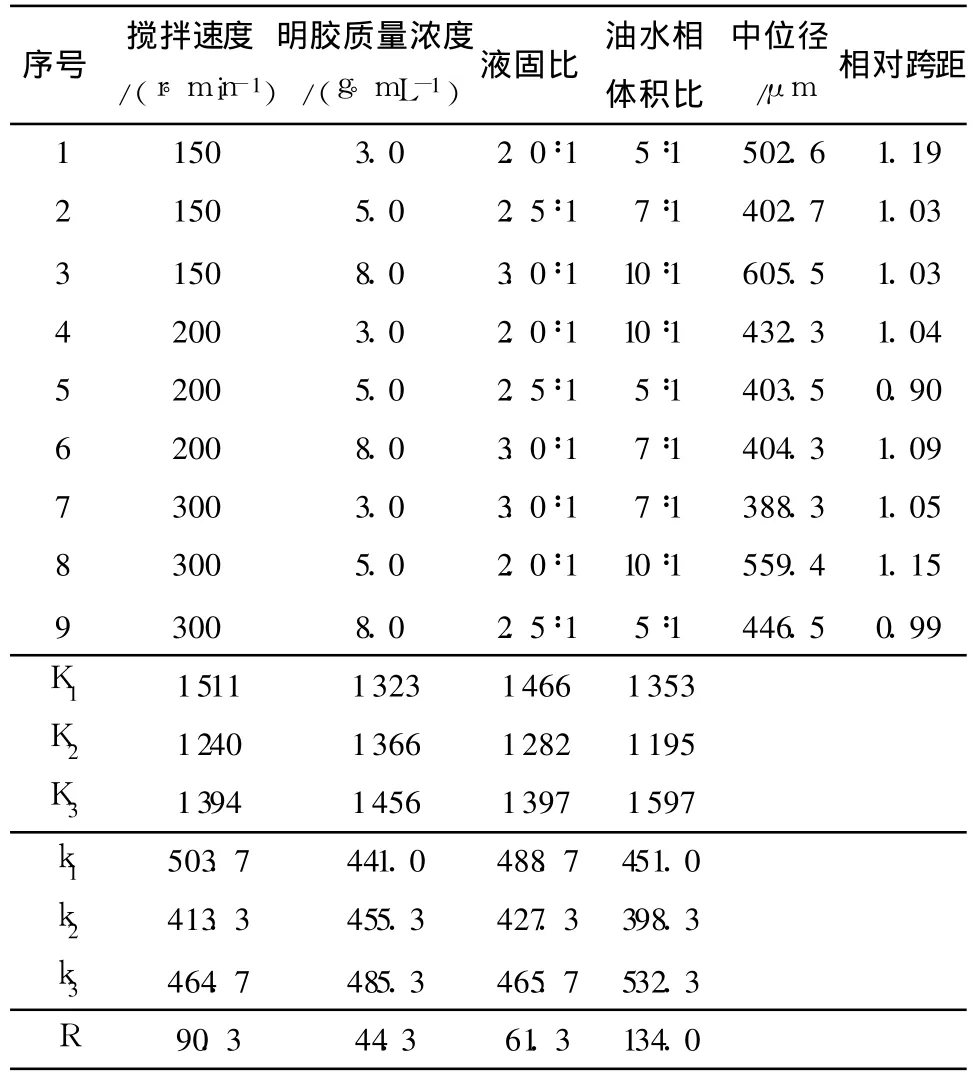
表2 L9(34)正交试验结果Table 2 Results of L9(34)orthogonal test
激光粒度仪测得微球的粒径约为 100~1000μm,中位径为 388~605μm,同时由表 2可见,相对跨距为0.90~1.19,这一范围与目前临床使用的磷酸钙微球的粒度范围一致.采用SPSS 16.0统计分析软件对实验数据进行分析,结果表明,各因素对微球粒径的影响程度为:油水相体积比 >搅拌速度 >液固比 >明胶质量浓度,其中油水相体积比对微球中位径的影响最大.分析发现,制备大粒径微球的实验方案为A1B3C1D3,按此工艺参数制备3批微球,微球的粒径分布如图 1所示.测得微球的中位径为(572.5±18.7)μm,相对跨距为 1.02±0.04,证明该方案重现性好,制备工艺可行.
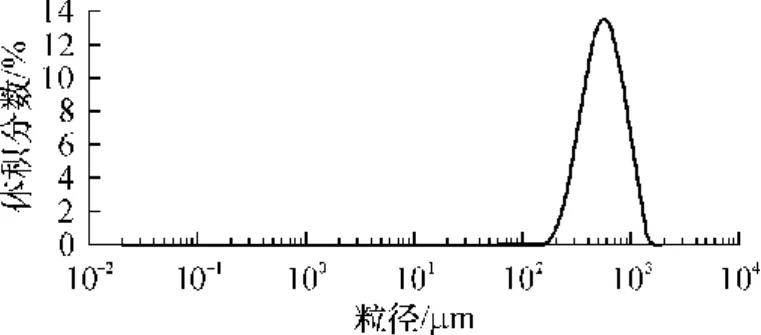
图1 自固化磷酸钙微球的粒径分布Fig.1 Size distribution of CPC microspheres
2.2 微球形貌和结构
自固化磷酸钙微球的SEM分析结果如图2所示.由图可见微球表面平整光滑,球形度良好,表面均匀分布有微小的孔洞.

图2 自固化磷酸钙微球的SEM照片Fig.2 SEM images of CPC microspheres
微球自然断面的SEM照片如图3所示.由图3可见,纳米针状的HA相互桥连形成团簇体,其边缘附有一层明胶,形成了明胶和针状HA团簇构成的三维骨架结构的杂化体,孔隙较多,形貌复杂.

图3 自固化磷酸钙微球断面的SEM照片Fig.3 SEM image of the cross section of the CPC microspheres
通过压汞法测得正交试验中 9组微球的孔隙率范围为43.4%~66.7%,测定了第4组和第6组微球内部开口孔的孔径分布,如图4所示.

图4 自固化磷酸钙微球的孔径分布Fig.4 Pore size distributions of CPCmicrospheres
由图4可见,微球的孔径集中在100nm~10μm之间,微球内部含有大量的胶凝微孔和较大孔,这种孔隙分布和孔隙率有利于后期对微球进行功能性修饰.
2.3 微球的物相组成
含添加剂的CPC微球、纯CPC水化固化体、反应原料PCCP及DCPA的XRD谱图如图5所示.与标准粉末衍射数据卡片(JCPDS∶01-074-0566)对照发现,CPC微球和纯CPC水化固化体的物相无明显差异,均为HA,峰形较为弥散,结晶程度较差,CPC微球的衍射峰中没有发现原料PCCP和DCPA的特征峰,表明水化反应完全,少量明胶和变性淀粉的加入不会明显影响HA的水化结晶过程[12].
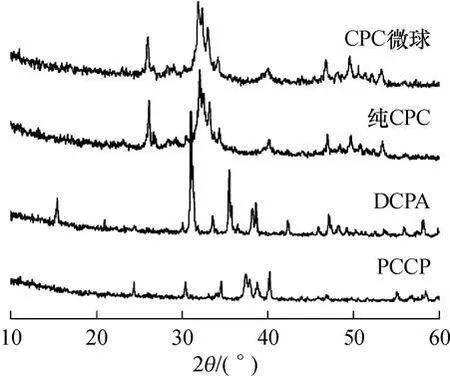
图5 各样品的XRD谱图Fig.5 XRD patterns of different samples
纯CPC水化样(不含添加剂)、CPC微球(含明胶)、CPC微球(含明胶和变性淀粉)的FT-IR分析结果如图6所示.
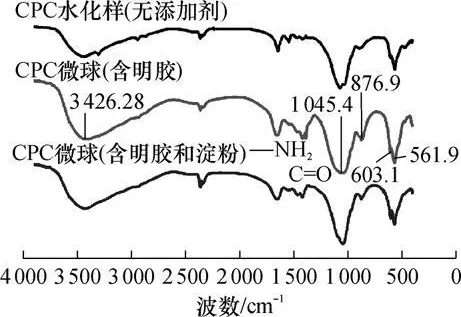
图6 含有添加剂和不含添加剂的CPC微球的FT-IR谱图Fig.6 FT-IR spectra of CPCm icrospheres with orwithoutadditives
由图 6可见,三者都具有典型的羟基磷灰石结构,在3426cm-1处有强收峰,处有曲振动峰,1045 cm-1处有缩振动峰,而明胶和变性淀粉分子链上的氨基(1540、1650cm-1)和羰基 (1300~1450cm-1)在两组微球样品中也有所体现.FT-IR结果从侧面证实了XRD分析结果,表明该配方的微球完全水化后的主要物相为弱结晶的 HA,而添加剂的加入不会影响磷酸钙的水化结晶过程.
2.4 微球的流动性和力学性能
休止角是表征粉体流动性的最常用方法之一,一般认为休止角 θ′≤30°时流动性好,θ′≤40°时可以满足生产过程中的流动性需求[13].用固定漏斗法测得微球的休止角为28.4°~31.6°,表明微球的流动性良好.
CPC微球的水化样和冷干样 (用冰晶冷冻干燥的方法阻止CPC颗粒的水化反应)的应力-应变曲线如图7所示.通过比较应力 -应变曲线可定性分析微球强度的相对大小.
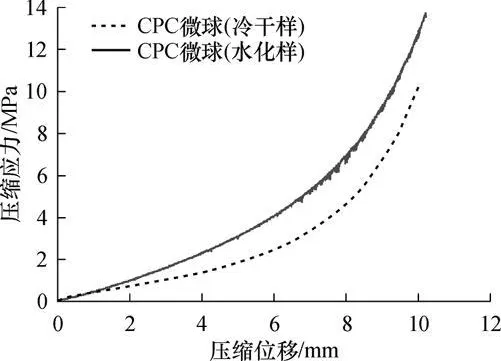
图7 CPC微球的应力-应变曲线Fig.7 Stress-strain curves of CPCm icrospheres
由图7可知,CPC微球经水化后强度有明显的提高,在压缩位移为6mm时,水化样的压缩应力提高了约40%.整个过程应力连续单调增大,表明由于含粘结剂,微球韧性良好,在受力过程中产生微球塑性形变,这一性质可使微球在临床手术操作中不易破碎.另外,水化微球的应力-应变曲线出现锯齿现象,表明水化的CPC微球变脆,压缩过程中发生了局部的开裂或破碎.
2.5 微球抗崩解性
在摇床中振荡3h后,c组HA微球出现浑浊,表明HA微球崩解;振动6h后,a组出现浑浊,表明不含变性淀粉的CPC微球开始崩解,此时b组液体依然保持澄清,未出现崩解;振动12h后,b组才浑浊,发生部分崩解.上述现象表明,CPC微球的抗崩解性要明显好于 HA微球,变性淀粉的加入可明显增强CPC微球的抗崩解性.
溶胶态的明胶具有良好的亲水性,与 HA颗粒亲和性较好[14].当用明胶作为粘结剂制备HA微球时,由于HA较难溶,其固化主要依靠明胶的胶结包裹作用.当 HA/明胶浆体受急冷时,处于溶胶状态的明胶立刻发生溶胶-凝胶转化而固化,HA颗粒被凝胶态的明胶包裹形成微球.但HA颗粒只是通过明胶结合在一起,微球容易因明胶在水溶液环境下的溶胀而迅速发生崩解.
CPC微球的成球除因明胶固化而产生的粘结和包裹作用外,其中的CPC在成球和后期的养护过程中,发生了充分的水化反应,通过一系列物理化学作用而进一步结合、固化.PCCP与DCPA在水化过程中,转化为大量相互桥连的纳米针状 HA的团簇[15],这一作用力大大增强了体系内部无机颗粒之间的结合,同时明胶和淀粉贴附包裹在HA团簇的表面,形成许多微孔和凝胶孔,构成一个复杂的三维结构的纳米HA团簇/明胶膜的杂化体,其形貌和周围的孔径分布如图3所示.除此之外,因PCCP和DCPA颗粒的溶解,溶液中产生大量的 Ca2+和阴离子基团,它们与明胶及淀粉分子链上的基团发生的复杂的作用,如Ca2+与变性淀粉中的阴离子发生螯合作用、淀粉链在CPC颗粒上发生的桥连作用以及淀粉与 CPC颗粒的静电引力作用等[16].因此,即使CPC微球内部的明胶发生溶胀溶出后,其中的无机颗粒仍然能够结合在一起,大大提高了CPC微球的抗崩解能力.
2.6 微球的细胞相容性
新西兰大白兔的骨髓间充质干细胞在CPC微球组和CPC对照组上经不同培养时间后的成活、增殖情况如图8所示.
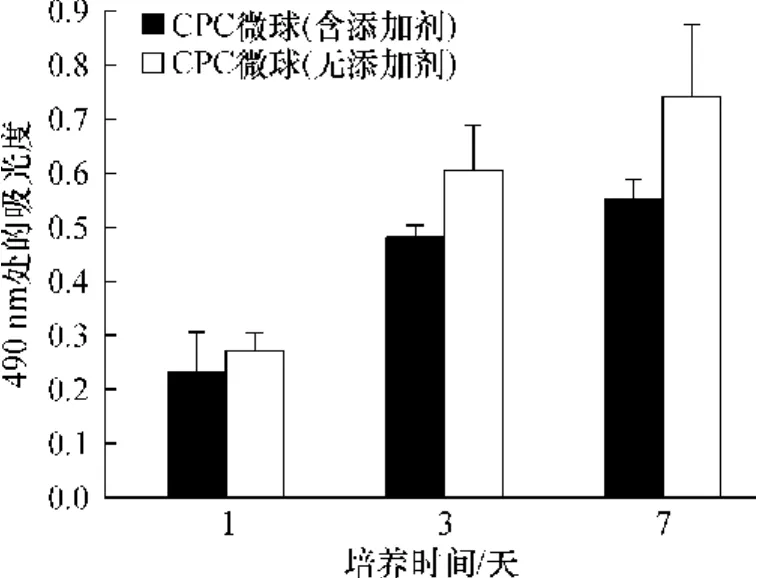
图8 细胞在自固化磷酸钙微球上的成活能力Fig.8 Cell viability on CPCmicrospheres
由图 8可见,随着培养时间的延长,吸光度增大,表明细胞的数量有所增加,细胞增殖情况属正常,其中细胞在无添加剂的CPC微球上的增殖略好于含添加剂的CPC微球.从细胞增殖情况来看,微球具有良好的细胞相容性.这一结果与人骨髓间充质干细胞在磷酸钙上的细胞毒性结果相吻合[17].
3 结论
(1)用反相乳液法制备了一种新型的自固化磷酸钙微球,制备的微球无需经过烧结.通过磷酸钙骨水泥的自固化、粘结剂的溶胶 -凝胶转化和变性淀粉的糊化及与磷酸钙颗粒产生的物理化学作用,使微球获得了较好的抗崩解性.
(2)所制备的自固化磷酸钙微球的粒径在 100~1000μm内.通过设计正交试验优化了制备工艺参数,当液固比为2.0∶1,搅拌速度为150 r/min、明胶质量浓度为8.0g/mL,油水相体积比10∶1时,可制备出中位径为572.5μm、粒度分布较窄的自固化磷酸钙微球,粒径分布范围能满足临床应用的要求.
(3)所制备的自固化磷酸钙微球表面光滑,流动性好,韧性和强度良好,便于充填、包埋等临床手术操作.细胞毒性试验表明这种CPC微球具有良好的细胞相容性.
[1] Misiek D J,Kent JN,Carr R F.Soft tissue responses to hydroxylapatite particles of different shapes[J].Journal of Oral and Maxillofacial Surgery,1984,42(3):150-160.
[2] Bodde EW H,Cammaert C T R,Wolke JG C,et al.Investigation as to the osteoinductivity of macroporous calcium phosphate cement in goats[J].Journal of BiomedicalMaterials Research:Part B,2007,83B(1):161-168.
[3] Ducheyne P,Hench L L,Kagan A,et al.Effect of hydroxyapatite impregnation on skeletal bonding of porous coated implants[J].Journalof Biomedical Materials Research,1980,14(3):225-237.
[4] Frame JW,Brady C L.Augmentation ofan atrophic edentulousmandible by interpositional grafting with hydroxylapatite[J].Journal of Oral and Maxillofacial Surgery, 1984,42(2):89-92.
[5] Councilon Dental Materials Instruments and Equipment, Councilon Dental Research,Council on Dental Therapeutics.Hydroxylapatite,beta tricalcium phosphate,and autogenous and allogeneic bone for filling periodontal defects,alveolar ridge augmentation,and pulp capping[J]. Journal of theMerican Dental Association,1984,108(5): 822-827.
[6] Zhang JP,Wang Q,Wang A Q.In situ generation of sodium alginate/hydroxyapatite nanocomposite beads as drug controlled releasematrices[J].Acta Biomaterialia, 2010,6(2):445-454.
[7] Bohner M,Gbureck U,Barralet JE.Technological issues for the development ofmore efficient calcium phosphate bone cements:a critical assessment[J].Biomaterials, 2005,26(33):6423-6429.
[8] Ye JD,Wang X P,Wang Y J.Rheological properties of an injectab le calcium phosphatebone cementand their relationship with the phase evolution[J].Key Engineering Materials,2007,336/337/338:1658-1661.
[9] Chen A Z,Li Y,Chau F T,et al.Application of organic nonsolvent in the process of solution-enhanced dispersion by supercritical CO2to prepare puerarin fine particles [J].Journal of Supercritical Fluids,2009,49(3):394-402.
[10] 蔡舒,姚康德,李鸿祥.羟基磷灰石合成及高温稳定性的研究[J].无机材料学报,2003,18(4):807-812.
Cai Shu,Yao Kang-de,Li Hong-xiang.Synthesis of hyd roxyapatite powder and its thermal stability[J]. Journal of Inorganic Materials,2003,18(4):807-812.
[11] Telli C,Serper,A,Dogan,A L,et al.Evaluation of the cytotoxicity of calcium phosphate root canal sealers by MTT assay[J].Journal of Endodontics,1999,25(12): 811-813.
[12] Chen L,Xiang H,Li X L,et al.Improvement of antiwashout performance of calcium phosphate cement using modified starch[J].Key Engineering Materials,2007, 336/337/338:1628-1631.
[13] Jain S K,Awasthi A M,Jain N K,et al.Calcium silicate based microspheres of repaglinide for gastroretentive floating drug delivery:preparation and in vitro characterization[J].Journal of Controlled release,2005,107 (2):300-309.
[14] Kim H W,Knowles JC,Kim H E.Hydroxyapatite and gelatin composite foams processed via novel freeze-drying and crosslinking for use as temporary hard tissue scaffolds[J].Journal of Biomedical Material and Research: Part A,2005,72A(2):136-145.
[15] Wang X P,Ye JD,Wang Y J.Hydrationmechanism ofa novel PCCP plus DCPA cement system[J].Journal of Materials Science:Materials in Medicine,2008,19(2): 813-816.
[16] Ambard A J,Mueninghoff L.Calcium phosphate cement: review ofmachanicaland biologicalproperties[J].Journal of Prosthodontics,2006,15(5):321-328.
[17] 温世锋,刘清华,郭奇峰,等.新型磷酸钙骨水泥与骨髓间充质干细胞的共培养研究 [J].临床医学工程, 2009,16(11):1-4.
Wen Shi-feng,Liu Qing-hua,Guo Qi-feng,et al.Research of co-culture with new injectable calcium phosphate bone cement and bone marrow-derived mesenchymal stem cells[J].Clinical Medical Engineering,2009, 16(11):1-4.
