金属氧化物负载纳米贵金属催化剂上的常温 CO氧化性能研究
2010-01-29廉红蕾潘维成
廉红蕾,潘维成
(1.郑州大学化工与能源学院,郑州 450001;2.中州大学化工食品学院,郑州 450044)
常温 CO催化氧化在封闭体系内微量 CO消除、CO2激光器及气体传感器等方面具有较强的实用价值。金基催化剂以其良好的低温活性和抗水性而受到广泛关注[1-6],我们的前期研究表明,将金负载在不同金属氧化物上,在 25℃、相对湿度为 80%的条件下,表现出不同的 CO氧化性能[7]。但研究多局限于单组分氧化物载体及单一贵金属组分,对双组分氧化物载体及双贵金属体系的研究较少。本文采用不同方法制备了系列负载型金基催化剂,考察了以双组分金属氧化物 ZnO-Fe2O3为载体的 Au/ZnO-Fe2O3催化剂及添加第二组分贵金属的Au-Pt/ZnO催化剂上的常温 CO氧化反应,研究了载体组成及第二组分贵金属对 CO氧化性能的影响,以期进一步了解催化剂的组成和结构同催化性能的关系。
1.实验部分
1.1 催化剂的制备
(1)2%Au/ZnO-Fe2O3催化剂的制备:采用共沉淀法制备不同 Zn/Fe摩尔比的 Au/ZnO-Fe2O3催化剂。试剂为分析纯的硝酸锌 (Zn(NO3)2·6H2O)、硝酸铁 (Fe(NO3)3·9H2O)、碳酸钠 (Na2CO3)和氯金酸 (HAuCl4·4H2O)。催化剂中 Au/(Zn+Fe)=2/100(摩尔比)。在充分搅拌下,将一定浓度的氯金酸与不同摩尔比的硝酸铁和硝酸锌的混合溶液滴入碳酸钠溶液中,待沉淀完全并陈化 4 h后,用 40℃-50℃的去离子水洗涤至无 Cl-检出,抽滤,干燥,在 300℃的20%O2/Ar气氛中焙烧 4 h,即制得催化剂。
(2)2%Au-Pt/ZnO催化剂的制备:采用两种方法制备不同Au/Pt摩尔比的Au-Pt/ZnO催化剂。试剂为分析纯的硝酸锌、碳酸钠、氯金酸和氯铂酸 (H2PtCl6·6H2O)。催化剂中 (Au+Pt)/Zn=2/100(摩尔比)。(a)共沉淀法 A:将氯金酸、氯铂酸 (Au/Pt=1/0.1或 1/0.2)与硝酸锌的水溶液混合,在充分搅拌下将该混合液滴入碳酸钠的水溶液中,继续陈化 4 h,用 40℃-50℃的去离子水洗涤至无 Cl-检出,抽滤,干燥,在 240℃的 20%O2/Ar气氛中焙烧 4 h。所得催化剂记为 2%Au-Pt/ZnO(A)。(b)共沉淀法 B:将硝酸锌与氯金酸的水溶液混合,在充分搅拌下滴加入碳酸钠的水溶液中,然后再将氯铂酸的水溶液滴入形成的沉淀中 (Au/Pt=1/0.1),继续陈化 4 h。后处理同上,所得催化剂记为 2%Au-Pt/ZnO(B)。
1.2 催化剂的表征
用Lab XRD 6000型 X射线衍射仪测定催化剂的物相结构。Cu Kα,镍单色器,电压 40 kV,电流 30 mA。步长 0.020,停留时间为 0.1 s。BET比表面积在 Micromeritics ASAP 2010型比表面测试仪上进行,N2为吸附质,在液氮温度下吸附。在自制的程序升温还原装置上进行 H2-TPR表征。载气为 H2(5%)和 Ar(95%)的混合气,取 20 mg催化剂在 50℃下用氩气预处理 30 min,降到室温后,用载气吹扫至基线平行,然后以 10℃/min的速度进行程序升温还原。H2耗量采用 Shimadzu GC-8A型气相色谱仪 (热导池检测器)分析。
1.3 催化剂的活性评价
催化反应在固定床连续流动反应器中进行,催化剂用量0.5 g。反应气预先经过加湿器,使其组成为:V(CO)∶V(O2)∶B(H2O)=0.5∶10∶1.8,Ar为稀释气 ,总流量 100 ml/min,反应温度 25℃。产物用 Shimadzu GC-8A型气相色谱仪 (热导池检测器)分析。催化剂的稳定性 (寿命)为 CO转化率从开始时的 100%下降至 99%的反应时间。
2.结果与讨论
2.1 常温加湿条件下 2%Au/ZnO-Fe2O3催化剂的 CO氧化性能
图 1是不同 Zn/Fe比例的双组分氧化物载体 2%Au/ZnO-Fe2O3催化剂上的 CO氧化反应性能。由图可见,所有样品都具有一定的常温 CO氧化稳定性,但载体组成 (即Zn/Fe摩尔比)对催化剂的常温 CO氧化稳定性有明显影响。总体来讲,Zn/Fe≤1时,样品的稳定性不是很好;Zn/Fe比例在 1/0.1~1/0.05之间时,样品稳定性提高,其中 Zn/Fe=1/0.05样品的稳定性最好,CO转化率可在 1100 h内维持100%。
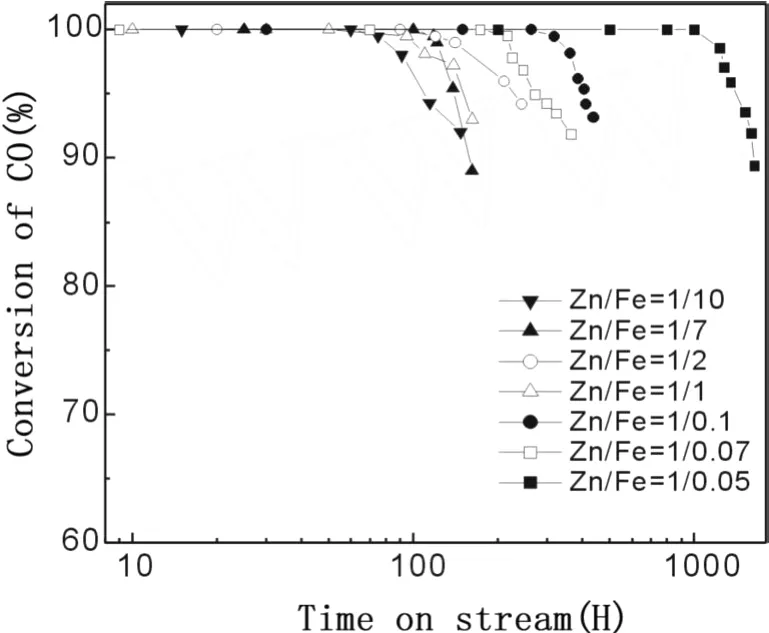
图 1 不同 Zn/Fe摩尔比的 2%Au/ZnO-Fe2O3催化剂上的 CO氧化反应性能
2.2 常温加湿条件下 2%Au-Pt/ZnO催化剂的 CO氧化性能
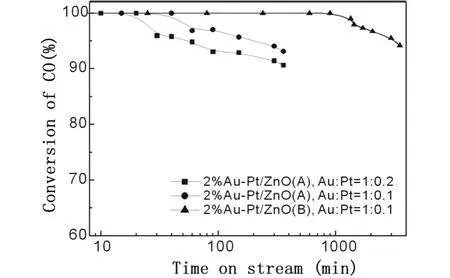
图 2 各种2%Au-Pt/ZnO催化剂上的 CO氧化反应性能
图 2是不同 2%Au-Pt/ZnO催化剂上的 CO氧化反应性能。由图可见,CO的初始转化率都能达到 100%,采用共沉淀法 A所制备的 Au/Pt=1/0.2和 1/0.1的 2%Au-Pt/ZnO(A)催化剂的寿命分别为 20和 40 min左右,随着反应的进行,CO转化率缓慢下降;在相同的反应时间内,Au/Pt=1/0.1的样品上 CO转化率更高一些。采用共沉淀法B制备的 2%Au-Pt/ZnO(B)催化剂上的 CO氧化反应稳定性可达900 min,高于共沉淀法 A所制备催化剂的稳定性。与我们前期报道的单一贵金属组分的金基催化剂 2%Au/ZnO相比[3,5],第二组分贵金属 Pt的添加明显降低了样品的常温CO氧化反应稳定性。
2.3 催化剂的 XRD表征
图 3(A)是不同 Zn/Fe摩尔比的 2%Au/ZnO-Fe2O3催化剂 CO氧化反应前的 XRD谱图。从图 3(A)(a-d)可以看到,当载体中铁的含量高于或等于锌的含量时,催化剂在2θ=30°~40°之间有一弱的宽峰,样品基本表现为无定型结构。图 3(A)(e-g)中,载体氧化物组成为 Zn/Fe=1/0.1~1/0.05,催化剂主要显示出氧化锌 (ZnO)的特征衍射锋,而且没有观察到单质金 (2θ=38.2°,44.4°和 65.5°)的特征衍射峰,表明在这些样品中金物种呈高分散状态。另外,不同催化剂的BET比表面积也有较大差异。
常温常湿 CO氧化反应后各 2%Au/ZnO-Fe2O3催化剂的 XRD谱图如图 3(B)所示。由图可见,当载体中铁的含量高于或等于锌的含量时 (图 3(B)(a-d)),反应后样品的XRD谱图与反应前相似,表现为无定型结构。对于 Zn/Fe=1/0.07和 1/0.05的样品,与 CO氧化反应前相比,反应后的样品除表现出 ZnO的特征衍射峰外,还出现了碱式碳酸锌Zn5(CO3)2(OH)6的特征衍射峰,表明随着反应的进行,在催化剂表面形成了类碳酸盐物种,这可能是引起样品活性下降的主要原因之一[3,8]。Zn/Fe=1/0.1样品反应前后的XRD谱图基本没有差别。
图 4是采用共沉淀法 A所制备的不同 Au/Pt比的 2%Au-Pt/ZnO样品 CO氧化反应前后的 XRD谱图。由图可见,反应前样品的 XRD谱图主要显示出 ZnO的特征衍射峰;反应失活后,出现碱式碳酸锌的特征衍射峰,而且在 2θ=38.2°,44.4°处出现了单质金的特征衍射峰,表明随着反应的进行,催化剂表面有类碳酸盐物种的累积,而且金粒子也逐渐发生聚集。与 2%Au/ZnO催化剂[3]相比,2%Au-Pt/ZnO催化剂稳定性不好的原因可能是 Pt物种的掺杂加速了类碳酸盐物种在反应过程中的累积,而且促进了金物种的聚集,从而引起样品上 CO氧化转化率在较短时间内就开始下降。
2.4 催化剂的 H2-TPR表征
对 2%Au/ZnO-Fe2O3(Zn/Fe=1/0.1~1/0.05)催化剂进行了 H2-TPR表征,结果如图 5所示。低温区域 (50℃~300℃)的还原峰可能与氧化金及载体中少量氧化铁的部分还原 (Fe2O3→Fe3O4)有关[6]。300℃以后的高温还原峰与载体 ZnO的连续还原[9]及掺杂的少量铁物种的还原 (主要是 Fe3O4→FeO→Fe)有关[6]。由图 5可见,不同 Zn/Fe比例样品的还原行为有较大差异,这可能是造成不同 Zn/Fe比例的催化剂常温常湿 CO氧化稳定性有较大差异的原因之一。
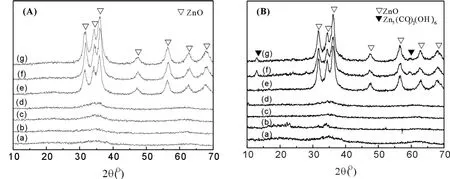
图 3 反应前 (A)和反应后 (B)的 2%Au/ZnO-Fe2O3催化剂的 XRD谱图(a)Zn:Fe=1:10;(b)Zn:Fe=1:7;(c)Zn:Fe=1:2;(d)Zn:Fe=1:1;(e)Zn:Fe=1:0.1;(f)Zn:Fe=1:0.07;(g)Zn:Fe=1:0.05
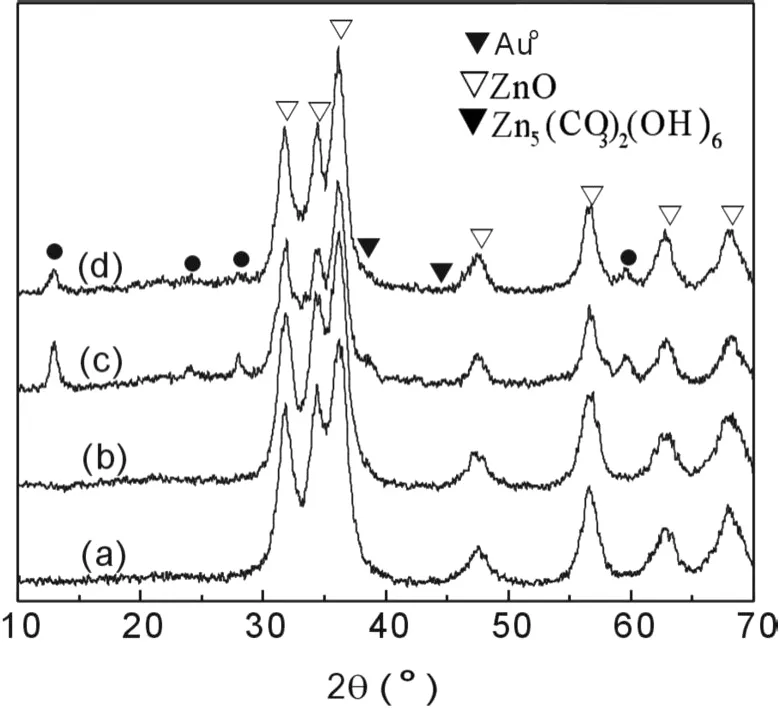
图 4 不同 2%Au-Pt/ZnO(A)催化剂的 XRD谱图(a)Au/Pt=1/0.1(反应前);(b)Au/Pt=1/0.2(反应前);(c)Au/Pt=1/0.1(反应后);(d)Au/Pt=1/0.2(反应后)
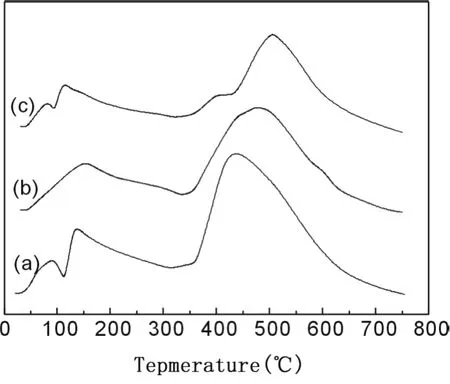
图 5 2%Au/ZnO-Fe2O3催化剂的 H2-TPR谱图Zn/Fe=1/0.1;(b)Zn/Fe=1/0.07;(c)Zn/Fe=1/0.05
对 2%Au-Pt/ZnO样品进行了 H2-TPR表征,结果如图 6所示。由图可见,在低温区,三个样品均在 156℃左右出现一还原峰,可归属为某种铂物种的还原,而且与 Pt/ZnO相比[9],铂的还原峰由 167℃位移至 156℃,说明在 Au-Pt/ZnO样品中,两种贵金属组分之间存在某种相互作用。另外,在 100℃附近还有一肩峰,可能与氧化态金物种的还原有关,其中共沉淀法 B制备的样品中 (图 6(c))该肩峰最明显。由图 6可以看出,不同样品中的贵金属组分,即反应的主要活性位的还原行为不同,说明不同 Au/Pt比例及不同制备方法制备的催化剂中有不同性质的活性位,这可能是引起样品上 CO氧化性能有差别的主要原因。300℃以后的还原峰可归属为 ZnO的连续还原,三个样品中载体的还原行为也有较大差别。
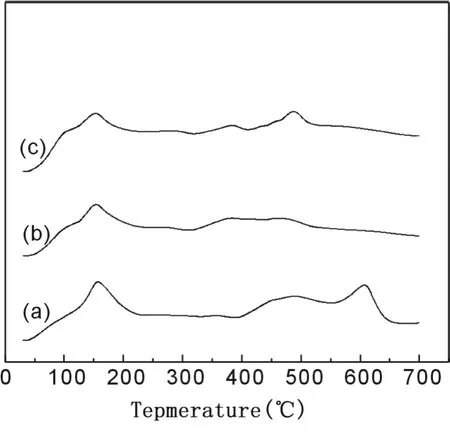
图 6 2%Au-Pt/ZnO催化剂的 H2-TPR谱图(a)Au-Pt/ZnO(A),Au/Pt=1/0.2;(b)Au-Pt/ZnO(A),Au/Pt=1/0.1;(c)Au-Pt/ZnO(B),Au/Pt=1/0.1
3.结论
通过对金属氧化物负载纳米贵金属催化剂上常温 CO氧化性能的研究可以得出:金属氧化物载体的组成对 2%Au/ZnO-Fe2O3催化剂的常温 CO氧化稳定性有较大影响,不同Zn/Fe比例催化剂的稳定性差别较大,其中 Zn/Fe=1/0.05样品的稳定性最好,可在 1100 h内维持 CO完全转化。不同载体组成的 2%Au/ZnO-Fe2O3催化剂的晶相结构、比表面积及氧化还原性质有较大差别。当 Zn/Fe≤1时,样品为无定型结构;而当 Zn/Fe=1/0.1~1/0.05时,催化剂主要显示出 ZnO的特征衍射锋;H2-TPR结果表明稳定性最好的 Zn/Fe=1/0.05样品中的金物种及部分氧化铁最容易被还原。第二组分贵金属 Pt的添加使 2%Au-Pt/ZnO催化剂在 CO氧化反应过程中容易发生类碳酸盐物种的累积及纳米金物种的聚集,从而引起常温 CO氧化反应性能下降。
[1]HarutaM,Tsubota S,Kobayashi T,et al.Low-Temperature Oxidation of CO over Gold Supported on TiO2,α-Fe2O3,and Co3O4[J].J Catal,1993,144(1):175-192.
[2]张文祥,陶玉国,贾明君,等.不同方法制备的纳米 Au/NiO上 CO常温常湿催化氧化性能的研究[J].高等学校化学学报,1998,19(8):1317-1319.
[3]Wang G Y,ZhangW X,Lian H L,et al.Effect of calcination temperatures and precipitant on the catalytic performance of Au/ZnO catalysts for CO oxidation at ambient temperature and in humid circumstances[J].Appl Catal A,2003,239(1-2):1-10.
[4]Lian H L,JiaM J,PanW C,et al.Gold-base catalysts supported on carbonate for low-temperature CO oxidation[J].Catal Commu,2005,6(1):47-51.
[5]Wang G Y,ZhangW X,Lian H L,et al.Effect of Au loading,H2O and CO concentration on the stability of Au/ZnO catalysts for room-temperature CO oxidation[J].React Kinet CatalLett,2002,75(2):343-351.
[6]Wang G Y,Lian H L,ZhangW X,et al.Stability and deactivation of Au/Fe2O3catalysts for CO oxidation at ambient temperature and moisture[J].Kinet Catal,2002,43(3):433-442.
[7]王桂英,张文祥,蒋大振,等.常温常湿条件下 Au/MeOx催化剂上 CO氧化性能 [J].化学学报,2000,58(12):1557-1562.
[8]王桂英,廉红蕾,徐跃,等.沉淀剂对 Au/ZnO催化剂上CO氧化性能及催化剂结构的影响[J].高等学校化学学报,2001,22(11):1873-1876.
[9]Ammari F,Lamotte J,Touroude R.An emergent catalyticmaterial:Pt/ZnO catalyst for selective hydrogenation of crotonaldehyde[J].J Catal,2004,221(1):32-42.
