液相色谱三级四极杆质谱法快速测定动物组织中维吉霉素M1残留量
2010-01-12陈小霞岳振峰叶卫翔赵凤娟饶丽葛丽雅欧阳姗
陈小霞,岳振峰,叶卫翔,赵凤娟,饶丽,葛丽雅,欧阳姗
1(深圳大学教务处,广东深圳,518060) 2(深圳出入境检验检疫局食品检验检疫技术中心,广东深圳,518067)
维吉霉素 (Virginiamycin),别名维吉尼亚霉素、维吉尼霉素、肥大霉素等。维吉霉素是美国史克药厂畜禽保健公司对链球菌发酵提取的抗生素。它由70%大环内酯 (M1)(图 1)和 30%环状多肽 (S1)(图2)混合组成。两者结构不同,抗菌范围也不同。维吉霉素的作用主要是抑制细菌的核糖体(Ribosome),从而阻止细菌蛋白质的合成而达到杀菌效果。维吉霉素可用于防治畜禽细菌性痢疾和鸡坏死性肠炎,改善饲料的利用率,促进畜禽的生长,已被广泛使用。但这类药物也存在一定的副作用,存在着潜在的食品安全风险。中国、美国、日本均规定了动物组织中维吉霉素的最高残留限量,其中以日本的限量规定最为全面和严格;欧盟则禁止维吉霉素作为饲料添加药物使用。维吉霉素在动物组织中主要以原形药物状态存在。目前各国法规中没有明确规定维吉霉素的残留标识物,但由于 M1为主要成分,国内外普遍将维吉霉素M1作为维吉霉素的残留标识物。
目前国内外关于动物源性食品中维吉霉素药物残留的检测方法研究报道均较少[1-13],方法包括琼脂扩散法[1]、酶联免疫法[2]、薄层色谱法与生物自显影联用法[3-4]、高效液相色谱法[5-10]和液质联用法[11-13],其中 GB/T 20765—2006规定了猪肝脏、肾脏、肌肉组织中维吉尼霉素M1残留量的测定方法,但该方法的线性范围未包括国内外的限量规定浓度,而且需要繁琐的固相萃取步骤,效率低下。本文采用乙腈脱蛋白同时提取目标物,水稀释,再利用液质联用法的选择性较高和抗干扰能力较强的特点,实现了动物组织样品中维吉霉素残留的快速测定和确证。

图 1 维吉霉素M1的分子结构
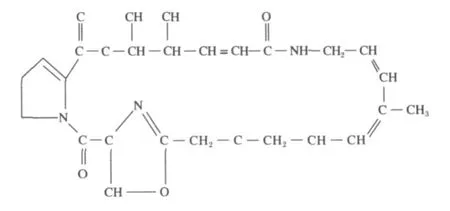
图 2 维吉霉素 S1的分子结构
1 实验部分
1.1 仪器与试剂
API3000型三级四极杆质谱仪 (美国应用生物系统公司),配电喷雾离子源和 Agilent 1100型液相色谱仪;MinishakerMS1型漩涡混合器 (美国 IKA公司);PT3000型均质器 (德国 Brinkmann公司);Turbo LV型吹氮浓缩仪 (美国 Zymark公司);Universal 32型低温离心机 (德国 Hettich公司);KQ-50B型超声清洗器 (昆山市超声仪器有限公司)等。
维吉霉素M1(Sigma公司,纯度约 95%);乙腈和甲酸均为色谱纯试剂;其他均为分析纯试剂;水为超纯水。
鸡肉、鸡肝、鸡肾、猪肾和牛肾等动物组织样品均为市购。
1.2 样品处理
称取试样 1 g(精确到 0.01 g),置于 15 mL具塞聚丙烯离心管中,加入 3 mL乙腈,旋涡混合 1 min,冰水浴超声提取 5min。以 3 000 r/min的转速在 15℃下离心 5 min,收集上清液于具有刻度的离心管中。离心后的残渣用 2 mL乙腈重复上述提取步骤 1次,合并上清液,用水稀释定容至 10mL,混匀。加入 4mL正己烷,混合 1min,3 000 r/min 5-15℃离心 5 min,弃去正己烷层,下层溶液过 0.22 μm滤膜后供液相色谱-质谱/质谱仪测定。
1.3 色谱质谱条件
Y MC-Pack Pro C18色谱柱 (3μm,100 mm ×2.0 mm i.d.);流速:0.3 mL/min;柱温:30℃;进样量:10 μL。流动相为乙腈和 0.003 mol/L甲酸铵缓冲液,洗脱梯度:乙腈的比例在 1.0min内由 30%线性提高到70%,保持 4.0min,柱平衡时间 5 min。
ESI+离子化模式;质谱分辨率:单位质量分辨率;监测模式:多反应监测 (MRM);雾化气:5.0 L/min;气帘气:12.0 L/min;喷雾电压:4.5 kV;去簇电压 (DP):50 V;聚焦电压 (FP):270 V;射人电压(EP):10 V;去溶剂温度:550℃;去溶剂气流:7.0 L/min;碰撞气 N2:6.0 L/min。其他条件见表 1。

表 1 维吉霉素M1的保留时间和优化质谱条件(带“*”号者为定量离子)
2 结果与讨论
2.1 质谱条件的优化
首先采用 10 mg/L的待测化合物的标准溶液以流动注射的方式在正离子模式下进行全扫描,确定分子离子,然后以待测化合物的分子离子为母离子,进行子离子扫描。选取丰度较强、干扰较小的 2个子离子为定性离子。最后以多反应监测 (MRM)正离子模式优化去簇电压 (DP)、聚焦电压 (FP)、射人电压(EP)、碰撞能量 (CE)、碰撞室射出电压 (CXP)、雾化气 (NEB)、气帘气 (CUR)、碰撞气 (CAD)、喷雾电压(IS)、离子化温度 (TEM)等各种质谱参数,得到最佳质谱条件 (表 1)。
2.2 色谱条件的优化
因维吉霉素M1分子结构中有多个氨基和羟基,能在水中发生解离,色谱柱固定相表面的残存硅醇基和金属离子可通过氢键或离子交换作用对其产生强烈吸附作用,出现色谱峰拖尾、保留时间不稳定或过长,甚至被保留在色谱柱上,导致峰形异常和分离度下降。本研究采用以高纯硅胶为基体并经端基封闭处理的 Y MC-Pack Pro C18柱进行了分离试验,发现该柱对维吉霉素和干扰成分具有良好的分离效果和对称的峰形,可满足质谱检测的要求。
国家标准 G B/T 20765—2006采用甲醇 +乙腈 (50+50)与 0.003mol/L甲酸铵缓冲液作为流动相,需要 35 min才能完成检测。本方法用乙腈代替甲醇 +乙腈(50+50),通过梯度优化,可将检测时间缩短到 10min,大幅提高了检测效率,而且峰形尖锐对称 (图 3)。
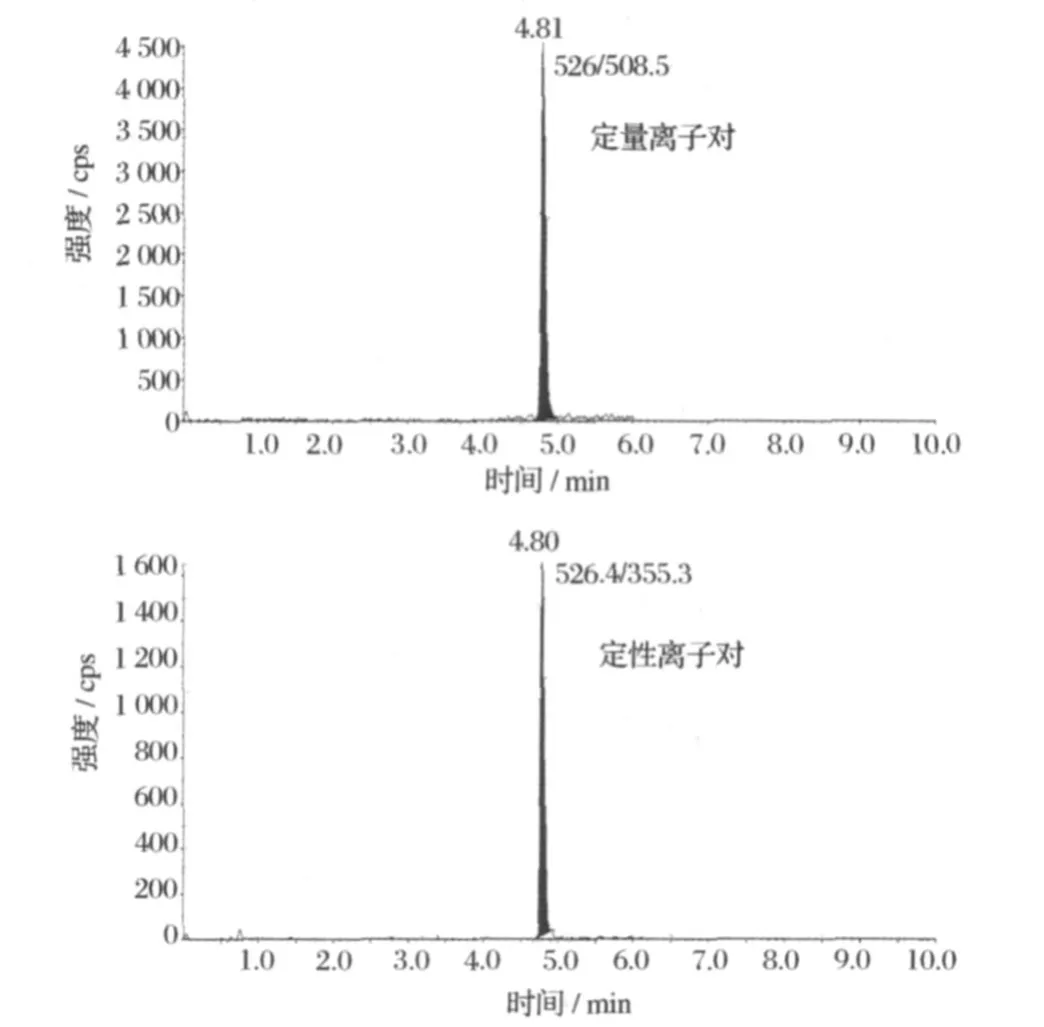
图 3 加标鸡肉样品中维吉霉素M1的多反应监测(MRM)色谱图 (加标浓度 25μg/kg)
2.3 提取和净化条件的选择
文献报道的提取溶剂主要有:甲醇 +磷酸氢二铵[5]、甲醇[6]、甲醇 +乙腈[7,12]、乙酸乙酯[9,11]、乙腈[13]等,本方法经比较试验发现,直接采用乙腈超声提取不仅可以减少脂肪等杂质的提出,而且回收率较高。文献报道的净化方法包括 C18固相萃取净化[1-3]、硅胶 +HLB固相萃取净化[4]等。本文经研究发现,样品提取液经水稀释后用正己烷净化,即可达到净化要求,不仅避免了烦琐的固相萃取净化步骤,而且降低了检测成本,提高了检测效率。
2.4 样品基质效应的考察
大气压喷雾电离离子源 (ESI)容易受样品基质的影响,但通过试验发现,基质匹配标准溶液与纯溶剂标准溶液的响应值不存在显著差异,因此维吉霉素M1不存在样品基质效应或样品基质效应不显著。
2.5 方法的线性关系和定量低限
在本检验方法所确定的实验条件下,取一系列标准溶液,以峰面积 (Y轴)对相应的维吉霉素M1的浓度 (X轴)作图,结果表明,标准溶液的浓度与对应的峰面积在 2-50μg/L内呈现良好的线性关系,线性方程为Y=2.96×103x+2.27×103,线性相关系数为0.999 5。经实验室内验证试验,确定本方法的定量低限为:鸡肉 25μg/kg、鸡肝 100μg/kg、鸡肾 100μg/kg、牛肾 100μg/kg、猪肾150μg/kg。
2.6 方法的回收率与精密度
以不含维吉霉素M1残留的动物组织为空白样品基质,综合考虑中国、美国、日本以及中国香港的最大残留限量规定,按照表 2进行 3个浓度水平进行添加回收试验,每个浓度水平进行 10次重复试验,测得维吉霉素的回收率和精密度汇总于表 3。本方法的回收率范围为:鸡肉 86.8%-108%、鸡肝 81.8%-109%、鸡肾 85.5%-102%、猪肾 82.7%-102%、牛肾 80.7%-103%,实验室内相对标准偏差为:鸡肉4.3% -6.7%、鸡肝 3.3%-7.3%、鸡肾 2.8%-4.6%、猪肾 1.9%-4.1%、牛肾 3.4%-6.9%,符合国内外对残留分析的要求。
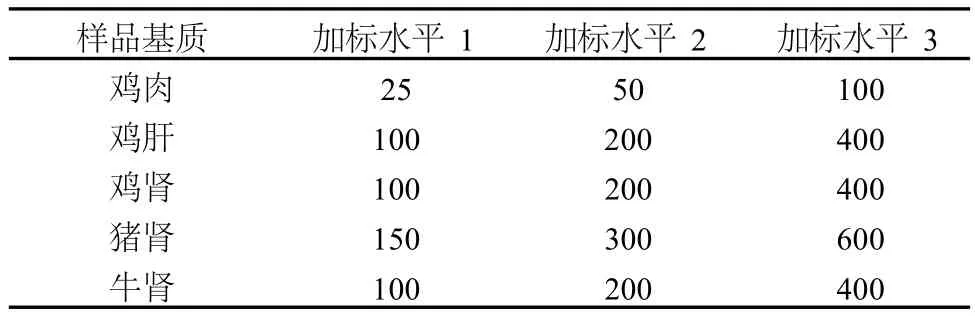
表 2 不同样品基质中维吉霉素M1的添加水平 μg/kg
2.7 实际样品分析
应用本方法分别检测鸡肉、鸡肝、鸡肾、猪肾和牛肾样品各 20批,检测结果统计情况汇总于表 4。可见,除猪肾和鸡肉样品各检出 1例不合格样品外,其他样品均合格。该方法具有灵敏高、选择性强、简便、快速等优点,方法的各项性能指标满足国内外食品安全法规的要求,可用于动物组织样品中维吉霉素M1残留的快速确证检测。

表 4 动物组织样品中维吉霉素M1残留的实际样品检测情况汇总表
[1] TsaiC E,Kondo F.I mproved agar diffusionmethod for detecting residual antimicrobial agents[J].Journal of Food Protection,2001,64(3):361-366.
[2] Situ C,Elliott C T.Simultaneous and rapid detection of five banned antibiotic growth promoters by immunoassay[J].Analytica Chimica Acta,2005,529(1/2):89-96.
[3] VincentU,Gizzi G,von Holst C,et al.Validation of an analyticalmethod for the determination of spiramycin,virginiamycin and tylosin in feeding-stuffs by thin-layer chromatography and bio-autography[J].Food Additives&Contaminants,2007,24(4):351-359.
[4] Gafner JL.Identification and semiquantitative estimation of antibiotics added to complete feeds,premixes,and concentrates[J].Journal ofAOAC International,1999,82(1):1-8.
[5] 耿志明,陈明,许大光,等.高效液相色谱法测定鸡组织中维吉尼亚霉素 M1的残留 [J].江苏农业学报,2005,21(3):172-175.
[6] 耿志明,陈明.高效液相色谱法测定饲料中维吉尼亚霉素M1的残留[J].江苏农业学报,2005,21(2):127-130.
[7] 耿志明,陈明,许大光,等.高效液相色谱法测定猪组织中维吉尼亚霉素 M1的残留 [J].中国兽药杂志,2005,39(2):10-13.
[8] Samanidou V F,Evaggelopoulou E N.Chromatographic analysis of banned antibacterial growth promoters in animal feed[J].Journal of Separation Science,2008,31(11):2 091-2 112.
[9] ChrisA J Hajee,Hans J A van Rhijn,Johan J P Lasaroma,et al.Development and validation of a method for the determination of sub-additive levels of virginiamycin in compound animal feeds by liquid chromatography[J].The Analyst,2001,126(8):1 332-1 338.
[10] Boulaire S,Bauduret J C,Andre F,et al.Veterinary drug residues survey in meat:an HPLC method with a matrix solid phase dispersion extraction[J].Journal of Agricultural and Food Chemistry,1997,45(6):2 134-2 142.
[11] 猪肝脏、肾脏、肌肉组织中维吉尼霉素M1残留量测定方法 液相色谱—串联质谱法 [S].GB/T 20765-2006.
[12] Boison J,Lee S,Gedir R.Analytical determination of virginiamycin drug residues in edible porcine tissues by LC-MSwith confirmation byLC-MS/MS[J].Journal ofAOAC International,2009,92(1):329-339.
[13] DellaW M Sin,Clare Ho,Wong Y C,et al.Simultaneous determination of lincomycin and virginiamycin M1in swine muscle,liver and kidney by liquid chromatography-electrospray ionization tandem mass spectrometry[J].Analytica Chimica Acta,2004,517(1-2):39-45.
