宏蛋白质组学:研究微生物群落的一种新策略
2010-01-12刘虎虎卢向阳
刘虎虎,田 云,卢向阳,方 俊
(1.湖南省农业生物工程研究所,湖南长沙 410128;2.湖南农业大学生物科学技术学院,湖南长沙 410128)
1 宏蛋白质组学的产生
微生物世界是分子多样性最大的天然资源库,由于地球上绝大部分微生物(约99%)是未培养微生物,即指那些利用分子生物学技术能够检测到,迄今所采用的微生物纯培养分离、培养方法还未获得纯培养的微生物。因此,传统的分离培养方法极大地限制了人们认识微生物世界的视野。由于未培养微生物在自然环境微生物群落中占有非常高的比例,无论是其物种类群,还是新陈代谢途径、生理生化反应、产物等都存在着不同程度的差异性和丰富的多样性,因而其中势必蕴涵着巨大的可开发生物资源。因此,如何有效开发利用这些微生物资源已受到广大科研人员的高度重视。随着生命科学与生物技术的飞速发展,多种分子生物学技术先后应用于微生物,特别是未培养微生物的研究,如利用16S rRNA分析复杂生态环境中未知微生物的种类。1994年,澳大利亚科学家W ilkins和W illiams[1]提出蛋白质组的概念,指一个基因组所表达的所有蛋白质,或细胞、组织或机体在特定时空所表达的全部蛋白质。蛋白质组学是研究一种细胞乃至一种生物所表达的全部蛋白质的生理结构与代谢过程的科学,但是对于组成复杂的天然样品或含有大量不可培养微生物的自然微生物区系,蛋白质组学研究还很不足。1998年,Handels man等[2]首次正式提出宏基因组的概念,最初用来定义土壤细菌混合基因组,研究对象是生境中全部微小生物遗传物质的总和(the genomes of the total microbiota found in nature),主要包括环境样品中的细菌和真菌。现在宏基因组是指特定环境中所有生物遗传物质的总和,以生态环境中全部DNA作为研究对象[3],避开了微生物分离培养的问题,极大地扩展了微生物资源的利用空间。目前宏基因组主要指环境样品中的细菌和真菌的基因组总和,也可指特定环境或共生体内所有生物遗传物质的总和,是一种不依赖于人工培养的微生物基因组分析技术[3]。而宏基因组学是一种以环境样品中的微生物群体基因组为研究对象,以功能基因筛选和测序分析为研究手段,以微生物多样性、种群结构、进化关系、功能活性、相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。宏基因组学的出现大大促进了对复杂微生物多样性及其遗传物质的研究,它在一定程度上弥补了传统研究方法所存在的局限性。宏基因组学的出现标志着基因组学分析从单一微生物扩展到复杂的微生物区系,但是它不能从基因水平上很好地解释系统的动态性。2004年,Rodriguez-Valera[4]在宏基因组和蛋白质组基础之上,进一步提出了宏蛋白质组的概念,它是指环境混合微生物群落中所有生物的蛋白质组总和。宏蛋白质组学则是针对某一特定地点的微生物群落所产生的全部蛋白质而进行大规模研究的方法[5]。宏蛋白质组学与宏基因组学在生物基因表达与功能水平上是相对应的,两者都是针对微生物特定时期动态的生命活动而进行的研究。两者对应关系[6]如图1所示。
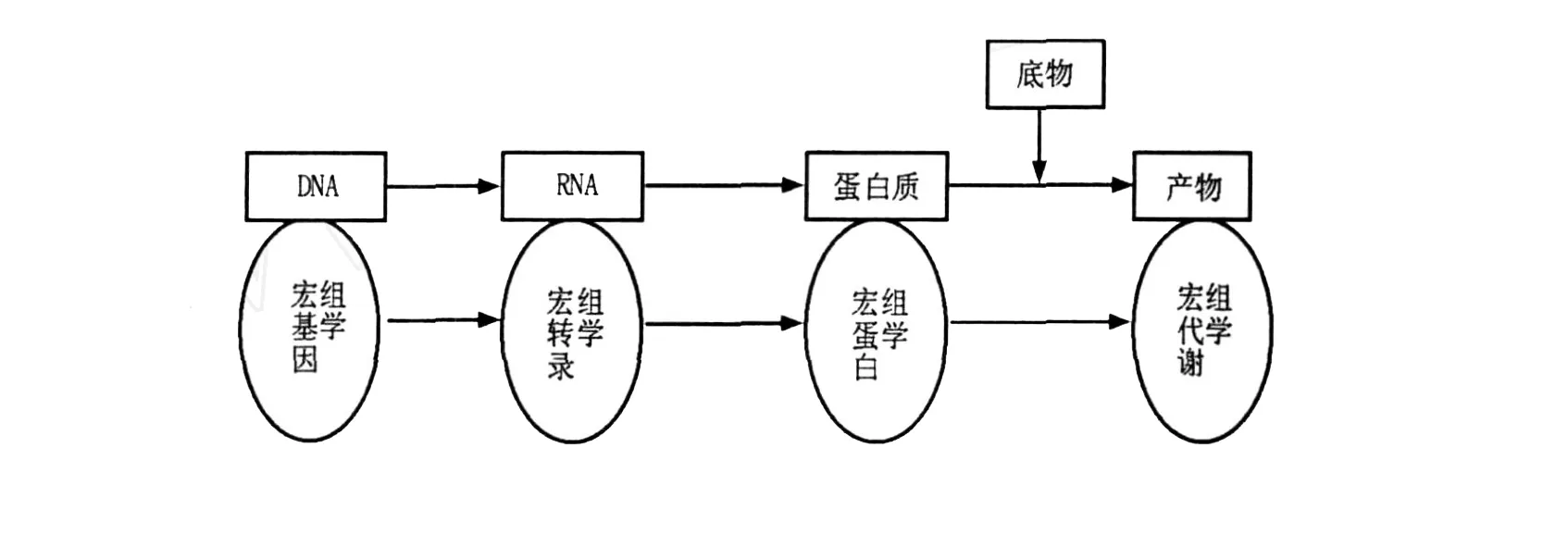
图1 微生物群落基因功能表达与研究示意图Fig.1 Schematic expression and research of gene in the ecology ofmicrobial communities
2 宏蛋白质组学研究策略及应用
2.1 宏蛋白质组学研究的基本技术路线及其策略
用于研究微生物群落的宏蛋白质组学与普通蛋白质组学在思路上基本相同。根据蛋白质理化性质的差异与不同的技术手段设计实验路线。首先,对环境混合微生物群落中所有生物的蛋白质进行提取与纯化。宏蛋白质组学的研究对象是特定环境中微生物所产生的所有蛋白质,因此,蛋白质的提取与纯化是宏蛋白质组学研究的关键,但目前尚未存在一种通用的方法用于对全部蛋白质的提取。2009年,Abram等[7]采用声波降解法与弗氏压碎器破碎法提取总蛋白质,并应用宏蛋白质组技术研究在污水处理过程中微生物厌氧消化时蛋白质的表达情况,实验结果表明,经声波降解法所提取的蛋白质能够增加双向电泳分离蛋白质效果;其次,对所有蛋白质进行分离。目前蛋白质的分离一般采用双向电泳与色谱技术;最后对蛋白质进行鉴定与分析。对于蛋白质的鉴定主要依赖于质谱技术。比较常用的质谱技术包括基质辅助激光解析电离-飞行时间质谱(Matrix-assisted laser desorption ionization time-of-flight mass spectrometry,MALD I-TOF-MS)、电喷雾质谱(Electrospray ionisation mass spectrometry,ESI-MS)与四极杆-飞行时间串联质谱(Quadrupole t ime-of-flight mass spectrometry,Q-TOF-MS)等。将经过质谱技术所鉴定的蛋白质序列输入数据库进行检索即可获得目的蛋白的相关信息,但目前蛋白质数据库中的蛋白质大多来源于已培养微生物,数据库中包含的未培养微生物蛋白质的信息非常少,这是现在宏蛋白质组学研究面临的一个重大问题[8]。宏蛋白质组学研究的基本技术路线如图2所示[9]。W i lmes提到宏蛋白质组学的研究主要有两个策略:一个是基于双向电泳技术分析鉴定微生物蛋白质表达;另一个则是基于液相色谱技术分析鉴定微生物蛋白质表达[10]。
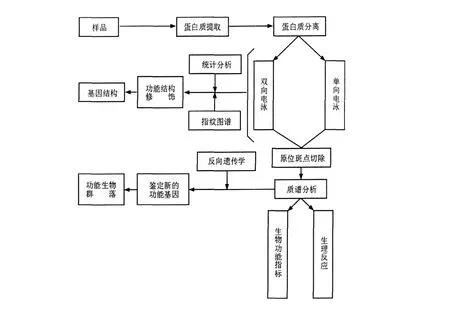
图2 宏蛋白质组学研究基本技术路线示意图Fig.2 Schematic approach of metaproteomics technique
2.2 宏蛋白质组学的应用
2.2.1 宏蛋白质组学在活性污泥微生物研究中的应用 Wilmes等[11]运用宏蛋白质组学技术研究活性污泥系统在强化生物除磷(Enhanced biological phosphorus removal,EBPR)过程中微生物蛋白质的表达情况。活性污泥在厌氧环境与好氧环境交替进行过程中可以增强生物除磷,在该过程中活性污泥微生物的蛋白质发挥着重要作用。经双向电泳和质谱技术鉴定出3个差异蛋白质,进一步通过数据库搜索和生物信息学分析推测这些蛋白质可能由β-变形细菌合成。W ilmes等[12]进一步运用双向电泳与荧光原位杂交技术研究不同处理方法对活性污泥微生物总蛋白质表达的影响,经反应器处理的活性污泥定义为“EBPR”样品,未经反应器处理的活性污泥定义为“nEBPR”样品,反应器可使活性污泥中微生物在厌氧环境与好氧环境不同两相中交替进行生物除磷。实验结果证明经反应器处理的“EBPR”样品分离得到630个蛋白质,反应前后存在表达差异的蛋白质占9.4%;而未经反应器处理的“nEBPR”样品分离得到590个蛋白质,存在表达差异的蛋白质占14.7%。在此基础之上,W ilmes等[13]运用双向电泳与质谱技术鉴定活性污泥中微生物在生物除磷过程中发挥重要作用的蛋白质,共鉴定出与生物除磷过程相关的蛋白质46个,这些蛋白质可能分别参与糖原的合成与分解、三羧酸循环、脂肪酸的合成等生化反应。Park等[14]利用宏蛋白质组学技术系统研究活性污泥蛋白质在厌氧与好氧等不同环境中的表达情况,并运用液相色谱-串联质谱法(Liquid chromatography-mass spectrography,LC-MS/MS)对差异蛋白质进行鉴定与分析,发现其中1个差异蛋白质为巴氏甲烷八叠球菌甲基化辅酶M还原酶(Methyl-coenzyme M reductase,MCMR)的亚基。
2.2.2 宏蛋白质组学在微生物起源研究中的应用 Kan等[15]利用双向电泳和LC-MS/MS对切萨皮克海湾不同水域中微生物群落进行研究,发现该海湾不同水域所分离的微生物蛋白质的表达存在差异,经质谱鉴定表明这些差异蛋白质与已知数据库的蛋白质不匹配,经进一步鉴定与生物信息学分析,其中3个蛋白质(CB1、CB3、CB6)可能与海洋微生物起源有关,其中CB3可能是NADH-辅酶Q氧化还原酶的亚基,而CB6与氨肽酶功能相类似。
2.2.3 宏蛋白质学在微生物胁迫应答中的应用Lacerda等[16]运用双向电泳、MALD I-TOF-MS与肽端从头测序技术研究重金属镉对细菌生物群落蛋白质表达的影响,结果发现重金属镉处理0.25 h与3 h时微生物的蛋白质表达发生显著变化,一共鉴定出109个差异表达蛋白质,这些蛋白质包括ATP酶、氧化还原酶和转运蛋白等,这是首次应用从头测序技术在大规模范围内研究未知微生物蛋白质的动态表达情况。
2.2.4 宏蛋白质组学在肠道微生物研究中的应用 Klaassene等[17]利用双向电泳与质谱技术分析了一个家庭中婴儿A和婴儿B在断奶之前不同时间排泄物中未培养微生物的生长情况,研究发现排泄物在不同时期所含微生物的蛋白质表达存在着一定的差异。研究以婴儿A 41 d排泄物为样品,应用MALD I-TOF-MS鉴定55个蛋白点并发现其与数据库不匹配。对于婴儿A不同时期(8、24、41 d)相同的4个蛋白质位点(1、2、7、11)进行鉴定分析发现其与已报道蛋白质存在差异。与此同时,研究发现1个含有肽端序列的蛋白质与双歧杆菌的转移酶相类似。Verberkmoes等[18]利用宏蛋白质组学技术研究幼儿双胞胎各自排泄物中蛋白质的表达情况,并以此推测人体肠道微生物的代谢情况,结果发现宏蛋白质组学相对于宏基因组学呈非正态分布。此外,研究发现样品中部分蛋白质属于同源蛋白,它们可能参与人体免疫和微生物代谢活动。实验之所以选择人体排泄物作为样品,是因为排泄物具有非侵入性,而且样品中部分蛋白质经鉴定分析与人体内蛋白质相同,这些蛋白质具有一定的代表性。
2.2.5 宏蛋白质组学在口腔微生物研究中的应用 J.D.Rudney等[19]应用三维肽段分离技术与串联质谱技术对分别取自口腔癌患者与正常人群唾液样品微生物进行宏蛋白质组分析。实验主要以口腔癌患者唾液为研究对象,并根据蛋白质理化性质差异分离鉴定出7 000个多肽,其中357个多肽与微生物起源相关。研究通过系统进化方法分析这些多肽发现其主要属细菌类群。在种分类学划分水平,仅鉴定出11%多肽;在门分类学划分水平,29%多肽属厚壁门,其中大部分多肽属链球菌属;20%多肽属变形菌门;4%多肽属放线菌门;3%多肽属拟杆菌门;1%多肽属螺旋体门等。研究经通路分析技术发现同源蛋白质簇(Clusters of Orthologous Groups,COGs)中37%多肽属J组,参与蛋白翻译;19%多肽属G组,参与糖酵解过程;8.2%多肽属E组,参与氨基酸代谢;7.9%多肽属C组,与产能有关。此外,研究还发现包括蚜虫内生共生体、极端微生物与古细菌在内的部分新物种。
2.2.6 宏蛋白质组学在膜蛋白研究中的应用RobertM Morris等[20]应用串联质谱技术对南大西洋10个不同区域样品表层水膜蛋白进行研究分析。通过构建16S rRNA文库并对849个16S rRNA序列比对分析发现其中184个序列属放线菌;31个序列属细菌浮游生物;90个序列属绿球藻;177个序列属SAR11。它们的分布情况反映出不同海域富含不同的营养物质。作者对来自10个样品共5 389个肽段进行鉴定并对939个特殊肽段作进一步分析,发现这些肽段大多属TBDTs(TonB-dependent transporters)、孔蛋白与ABC转运蛋白。这些蛋白质主要参与物质运输过程,其中TBDTs通过质子动力势转运营养物质。研究采用量多元尺度法与串联质谱技术联合分析发现不同蛋白在沿海水域与公海水域含量存在差异。同时,研究发现不同水域样品中均含有视紫红质并推测光营养细菌浮游生物可以利用光能参与不同形式的生命活动。此外,在南大西洋上层流发现古细菌氨单加氧酶与病毒蛋白,据此推测古细菌作为一类重要的硝化细菌在富含营养的表层水中起着重要的作用。
3 存在问题与展望
宏基因组学为微生物资源的开发利用提供了重要的信息,同时也为未培养微生物的研究提供了新的技术手段,但目前尚无较好方法用于研究微生物与环境之间的相互作用。宏蛋白质组学概念的提出为解决这一难题提供了可能,它以微生物群落所产生的所有蛋白质为研究对象,应用宏蛋白质组技术对其进行鉴定分析并研究微生物在复杂环境中生命活动的动态变化。宏蛋白质组学的研究工作目前尚处于初级阶段,仍存在许多问题有待解决。一方面来自微生物内因,地球上约99%微生物属未培养微生物,而且几乎所有的微生物在其生命周期中都会遇到各种不同的环境。这些微生物需要适当调整其所表达的蛋白质,从而维护细胞内部的代谢平衡。大多数微生物在不同的生长环境下仅表达它们整个蛋白质组中的一部分,较难获得环境样品中微生物所表达的高宏蛋白质组覆盖率;另一方面来自研究技术的局限性。比如双向凝胶电泳技术不能够检测低丰度蛋白或具有极端物理化学属性的蛋白,值得一提的是多维蛋白鉴定技术与GeLC-MS/MS方法在上述方面已取得一些进步。此外,用来将肽段与特异性蛋白进行匹配的生物信息学方法可能会产生假阳性鉴定结果,从而影响对于蛋白质的分析鉴定。因此,解决这些问题成为推动宏蛋白质组学技术发展的一个主要力量。随着科学技术的不断发展,宏蛋白质组学将在微生物群落研究中发挥越来越重要的作用。
[1] Graves PR,Haystead TA.Molecular biologist’s guide to proteomics[J].MicrobiolMolBiol Rev,2002,66(1):39-63.
[2] Handels man J,Rondon MR,Brady SF,et al.Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J].Chem Biol,1998,5:245-249.
[3] Rondon MR,August PR,Bettermann AD,et al.Cloning the soilmetagenome:a strategy for accessing the genetic and functional diversity of uncultured microorganis ms[J].Appl Environ Microbiol,2000,66:2541-2547.
[4] Rodríguez-Valera F.Environmental genomics,the big picture?[J].FEMS Microbiol Lett,2004,16:231(2):153-158.
[5] Wilmes P,Bond PL.The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms[J].Environ Microbiol,2004,6:911-920.
[6] Maron PA,Ranjard L,Mougel C,et al.Metaproteomics:a new approach for studying functional microbial ecology[J].Microb Ecol,2007,53:486-493.
[7] Abram F,Gunnigle E,O’Flaherty V.Optimisation of protein extraction and 2-DE for metaproteomics of microbial communities from anaerobic wastewater treatment biofilms[J].Electrophoresis,2009,30:4149-4151.
[8] 郝纯,刘庆华,杨俊仕,等.宏蛋白质组学:探索环境微生态系统的功能[J].应用与环境生物学报,2008,14:270-275.
[9] Maron PA,Ranjard L,Mougel C,et al.Metaproteomics:a new approach for studying functional microbial ecology[J].Microb Ecol,2007,53:486-493.
[10] Wilmes P,Bond PL.Metaproteomics:studying functional gene expression in microbial ecosystems[J].Trends Microbiol,2006,14:92-97.
[11] Wilmes P,Bond PL.The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganis ms[J].Environ Microbiol,2004,6:911-920.
[12] Wilmes P,Bond PL.Towards exposure of elusive metabolic mixed-culture processes:the application of metaproteomic analyses to activated sludge[J].Water Sci Technol,2006,54:217-226.
[13] Wilmes P,Wexler M,Bond PL.Metaproteomics provides functional insight into activated sludge wastewater treatment[J].PLoSOne,2008,3:1-11.
[14] Park C,Helm RF.Application of metaproteomic analysis for studying extracellular polymeric substances(EPS)in activated sludge flocs and their fate in sludge digestion[J].Water Sci Technol,2008,57:2009-2015.
[15] Kan J,Hanson TE,Ginter JM,et al.Metaproteomic analysis of Chesapeake Bay microbial communities[J].Saline Systems,2005,1:7.
[16] Lacerda CM,Choe LH,Reardon KF.Metaproteomic analysis of a bacterial community response to cadmium exposure[J].J Proteome Res,2007,6:1145-1152.
[17] Klaassens ES,de VosWM,Vaughan EE.Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract[J].Appl Environ Microbiol,2007,73:1388-1392.
[18] Verberkmoes NC,Russell AL,Shah M,et al.Shotgunmetaproteomics of the human distal gut microbiota[J].IS ME J,2009,3:179-189.
[19] Rudney JD,Xie H,Rhodus NL,et al.A metaproteomic analysis of the human salivary microbiota by three-dimensional peptide fractionation and tandem mass spectrometry[J].Molecular Oral Microbiology,2010,25:38-49.
[20] Morris RM,Nunn BL,Frazar C,et al.Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction[J].The IS ME Journal,2010,4:673-685.
