2-氨基丙醇的合成研究
2010-01-11王伟强顾海宁李小玲
王伟强 顾海宁 李小玲
(1.浙江京新药业股份有限公司,浙江 新昌 312500;2.浙江大学分析测试中心,浙江 杭州 310028;3.杭州师范大学,材化学院,浙江 杭州 310036;4.杭州广林生物医药有限公司,浙江 杭州 310028)
0 前言
喹诺酮类抗菌药具有抗菌谱广、抗菌活性强、给药方便、不良反应小、与其他抗生素无交叉耐药性等优点而成为临床联合用药的首选,用量已超过头孢类抗生素,成为第一大抗菌用药。2-氨基丙醇,作为合成喹诺酮类抗菌药氧氟沙星[1]的起始原料,其合成尤显重要。
1 合成路线研究
关于2-氨基丙醇的合成,国内报道[2、3]较少。课题组结合已有报道的(S)-(+)-2-氨基丙醇合成工艺,对2-氨基丙醇的合成方法进行了探索研究。
1.1 羟基丙酮氨解法
蒋锦等[2]课题组采用氯丙酮为原料,以碳酸二甲酯为溶剂、KI为催化剂,经乙酸基化后,水解得到羟基丙酮,再经Raney Ni催化下还原氨化制得目标产物1,总收率44%。反应式如下:
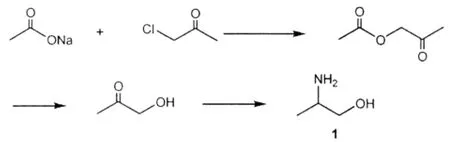

该方法所涉及到羟基丙酮成本较贵,催化剂制备复杂,产品收率不高,因此不适合工业化生产。
1.2 丙氨酸还原法[4~6]
此类方法是以丙氨酸或丙氨酸衍生物为起始原料,经Raney Ni、NaBH4等还原剂还原得到2-氨基丙醇。这类方法所用到的原料和还原剂价格昂贵,不宜大规模生产。
1.3 2-氯-1-丙醇氨解法
方法以环氧丙烷与盐酸开环生成2-氯-1-丙醇,然后以2-氯-1-丙醇为原料氨解制得2-氨基丙醇,反应式如下:
该工艺解决了上述工艺原料成本过高的问题,而且生产工艺简单,操作条件相对温和,是一条全新的合成工艺,但是也存在不足之处,理论上环氧丙烷与盐酸开环加成主产物为2-氯-1-丙醇,即β-氯丙醇,而实验结果主产物却是α-氯丙醇,StewartC A等[7]人的实验结果显示β-氯丙醇和α-氯丙醇的摩尔比为11:89,国内张胜帮等[8]也报道了上述开环反应,结果显示主要产物也是α-氯丙醇。
1.4 新工艺方法
为解决上述第二种工艺方法中反应产物的选择性问题,课题组经过实验研究,采用精馏方法分别提取β-氯丙醇和α-氯丙醇,其中β-氯丙醇经催化氨解制得目标产物1;α-氯丙醇经皂化反应生成起始原料环氧丙烷,如此循环进行:

在环氧丙烷的开环反应中,通过对反应机理的探讨,课题组认为该反应是一个符合马式规则的烯烃亲电加成反应,即HO-Cl中的卤素正离子大部分加成到带有较多氢原子的碳上,形成α-氯丙醇。
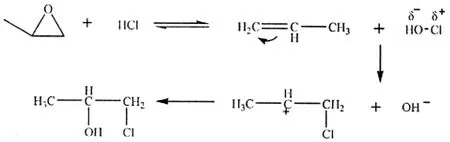
为提高转化率,并使反应得到尽可能多的β-氯丙醇,课题组对反应的温度、投料方式和反应溶剂进行了优化:控制反应温度在55~60℃,采用无溶剂状态下,直接滴加环氧丙烷到液面下的投料方式,减少了原料的损失,使得转化率提高到了92.7%,β-氯丙醇和α-氯丙醇的摩尔比提高到了40:60。
该方法是对第二种工艺方法的有效改进,不仅降低了成本,而且提高了原子经济性、减少了三废排放量,适合工业化生产。
2 实验部分
2.1 仪器及试剂
仪器为Bruker-400MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);GC-16A型气相色谱仪、Bruker Vector22型红外光谱仪(Shimadzu公司,日本);化学试剂均为化学纯。
2.2 合成部分
2.2.1 β-氯丙醇和α-氯丙醇的制备
370g浓盐酸加到80g水中配成30%的盐酸水溶液,预热到50℃,开始滴加冷的200g环氧丙烷,直接滴加到液面下,滴加时温度维持在55~60℃,约45min滴加完毕,搅拌15min,冷却至室温,加固体无水碳酸钠中和反应液至pH=7,静置分层,水层待用,上层油层用无水硫酸钠干燥后过滤,滤液进行精馏,收集到127~130℃馏分即α-氯丙醇180.1g,132~134℃馏分即β-氯丙醇122.0g。
2.2.2 α-氯丙醇的循环利用
将85g氢氧化钙与100g水混合成石灰乳悬浮液,搅拌预热至100℃左右,另外将馏分α-氯丙醇180.1g加到上述水层中,搅拌预热至90℃,然后滴加到悬浮液中,滴加时保持反应液温度在95~100℃,同时蒸汽气提出环氧丙烷,约45min后反应结束,蒸出物冷却静置分层取上层即环氧丙烷层,滴加到50℃的盐酸水溶液(30%,250g)中,滴加时温度维持在55~60℃,约35min滴加完毕,搅拌15min,冷却至室温,加固体无水碳酸钠中和反应液至pH=7,静置分层取上层油层,用无水硫酸钠干燥后过滤,滤液进行精馏,收集到132~134℃馏分即β-氯丙醇65.0g。
循环反应一次,β-氯丙醇的总收率为57.4%。
2.2.3 2-氨基丙醇的制备
将30g β-氯丙醇和0.5g KI加到带搅拌器的高压反应釜中,用氮气置换釜内空气3次,然后通入过量的液氨(约100g),使釜内氨压约为0.8MPa,搅拌下逐渐加热升温,保持温度在130~140℃下反应约20h,停止反应,冷却至室温,放空氨气,滤除固体,减压蒸馏收集72~74℃/1.3KPa馏分,即2-氨基丙醇共18.5g,收率77.7%,GC含量>99%。
1H NMR(CDC13)δ:1.04(3H,d,J=6.4Hz,CH3),2.99(1H,m,CH),3.23,3.50(2H,m,CH2)。
3 结论
本工艺反应简单,操作方便;采用了循环反应的方法回收套用副产物,原子经济性高;避免使用昂贵的Raney Ni、NaBH4等作为还原剂,大大降低了成本,减少了污染物的排放,符合绿色化学的发展方向,工业化前景良好。
[1]杜 璇,屠锡德.氧氟沙星的临床用药进展[J].江苏药学与临床研究,2005,13(5):16-18.
[2]蒋 锦,王玉成,郭慧元.(R,S)-2-氨基丙醇的制备[J].中国医药工业杂志,2006,37(1):8-9.
[3]王菊仙,郭 强,郭慧元.2-氨基丙醇的制备[J].中国医药工业杂志,2007,38(8):551-556.
[4]AntonsS,BeitzkeB.Processforoptically active alcohols:US,5731479[P].1998-03-24.
[5]Antons S,Beitzke B.Process for optically active amino alcohols:US,6310254[P].2001-10-30.
[6]Jere F,Jackson J,Miller D.Kinetics ofthe aqueous phase hydrogenation of L-alanine to L-alaninol[J].Ind Eng Chem Res,2004,43:3297-3303.
[7]Stewart C A,Calvix A,Vaxder Werf.Reaction of propylene oxide with hydrogen halides.[J].J Am Chem Soc.1954,76:1259-1264.
[8]张胜帮,邵利民.环氧丙烷在盐酸中开环反应的机理[J].应用化学,2000,17(2):214-216.
