手性中间体氢化阿托酸的合成新法
2010-01-11李仲新斯剑锋申屠阳刘志滨
李仲新 斯剑锋 申屠阳 陈 芳 刘志滨
(1.浙江普洛医药科技有限公司,浙江 东阳 322118;2.浙江省东阳市环境监察大队,浙江 东阳 322118)
手性(chirality)一词是由希腊(Greece)语"手"衍生而来,即手征性的意思。人们的左右手互为镜像,不能以精确无误的相同方位将一只手直接放在另一只手之上。手性物质分子与其镜像就不可叠加,而非手性物质分子有可叠加的镜像。物质分子的手性是自然界的一种属性,同时展示着手性物质的独有的光学活性。
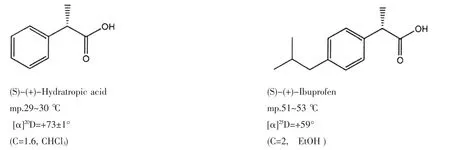
自然界中手性物质分子无所不在,氢化阿托酸(Hydratropic acid)就是一个例子,它最早是从天然产物马钱子碱(Strychnine)分离得到,后来又个天然产物金鸡纳霜(Quinine)分离物中获得,也有用天然产物麻黄素(Ephedrine)分离得到的扁桃酸(Mandelic acid),再经化学转化为标题物。上述几种天然产物也都是手性物质[1]。
20世纪初,Cushny发现天仙子胺(Hyoscyamine)的一个对映异构体比另一个的药效更大,其立体结构式如下:

从此就将手性与制药工业联系起来,当今大多数新药和发展中药物都存在着单一的光学活性异构体,手性中间体(Chiral intermediate)和相关产物约有80%进入制药行业[2],可见研究和合成手性中间体就直接关系到医药的发展。类似氢化阿托酸手性结构的化合物都具有消炎,镇痛和解热的功效,列举数例[2]:

标题物不仅是个手性中间体,手性子(chiral building block),而且还用作手性诊断剂(chiral diagnostic reagent)和拆分剂(Resolving agent)。类似的情况很普遍。手性化合物的合成,一般都是先制成其消旋物,然后再进行拆分。文献报导的标题物合成方法虽然较多,但是具有实用价值的却很少。较早的Eliel等[3]采用氯化试剂(亚硫酰氯或盐酸)以氯取代苯基乳酸的羟基,经甲酯化(或不酯化),再用氢化铝锂还原,只得到氢化阿托醇,收率较低,他们还采用硝酸银在碱性条件下氧化氢化阿托醛为相应的酸,收率74%。几乎在同时,Bonner[4]采用五氯化磷以氯取代苯基乳酸乙酯的羟基,再与苯硫酚反应,脱去氯化氢得到硫醚化合物,接着用Raney-镍还原脱去苯硫基,水解酯获得相应的酸,各步收率都在80%以上,他还用苯乙腈与碘甲烷/金属钠进行α-甲基化反应,水解氰基得到相应的酸,收率只有37%;Roger等[1]用氢化阿托醛与羟胺盐酸盐缩合得到相应的醛肟,再用乙酐脱水得相应的腈,水解腈为相应的酸,收率47%,在此后的15年,Angres[5]重复此法收率只41%,另外还有采用较为难得的化学试剂或较苛刻的反应条件来制取标题物的,就不再予以举例。
我们从实用性出发,设计出如下的合成路线制得标题物:

1 实验部分
1.1 试剂
工业级苯乙烯,自制干燥的氯化氢,30%的氰化钠水溶液(工业生产品),工业级氯仿,工业级乙醇,工业级无水碳酸钠,工业级盐酸
1.2 仪器
实验室用标准磨口玻璃仪器;
分析仪器:气相色谱仪GC-9790J。
1.3 实验操作
1.3.1 α-氯代乙基苯[II]
将104g(1.0mol)苯乙烯[I]溶解于150mL氯仿中加到反应瓶中,冷却至0℃,再向体系内慢慢通入干燥的氯化氢气体,在反应过程中要控制好温度,并且要适时取样分析,确认反应进程,直至反应基本完成,停止通气,加入适量无水碳酸钠粉,搅拌片刻,用以除去反应溶液内过量的氯化氢,过滤除去碳酸钠,蒸馏回收溶剂氯仿,减压蒸馏产物[II](82°/20mm或72°/16mm,48°/10mm)得130g左右,收率:90%~95%,b.p.194~195℃(分解)。

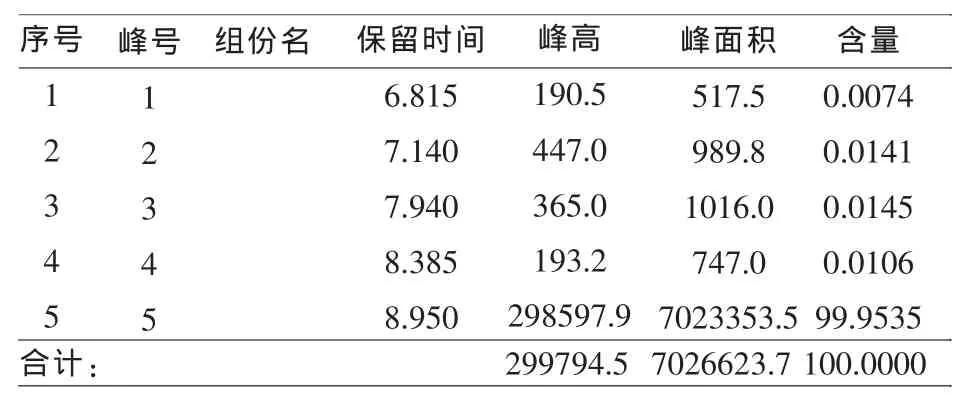
序号12345峰号12345组份名 保留时间6.815 7.140 7.940 8.385 8.950峰高190.5 447.0 365.0 193.2 298597.9峰面积517.5 989.8 1016.0 747.0 7023353.5含量0.0074 0.0141 0.0145 0.0106 99.9535合计: 299794.5 7026623.7 100.0000
1.3.2 氢化阿托腈[III]
将[II]130g溶解于200mL 95%乙醇中,并加到反应瓶内,另将170g 30%氰化钠水溶液加到滴液漏斗内,在搅拌下加热反应体系至60~70℃,开始滴加氰化钠水溶液,同时要控制好反应体系pH为6左右,当滴加完后,再继续反应1~2h,取样分析,至反应完全,冷却至室温,过滤不溶的无机盐,蒸馏回收溶剂乙醇并夹带部分水,计量回收差不多时,冷却,过滤不溶物,加入适量氯仿,搅动,使水相与有机相分开,取出水相,再用少量氯仿提取水相1~2次,将提取氯仿与有机相合并,干燥,蒸馏回收有机溶剂,在减压下106~108℃/12mm或74℃/0.5mm蒸出产物[III]约100g左右,收率:80%~90%。


序号123456峰号123456组份名 保留时间2.450 2.520 2.6050 3.100 6.685 10.760峰高587.3 2141.3 1514.3 243.0 403.3 59318.7峰面积903.1 2668.2 1880.9 383.2 1000.3 1569095.0含量0.0573 0.1693 0.1194 0.0243 0.0635 99.5662合计: 64207.9 1575930.7 100.0000
1.3.3 氢化阿托酸[IV]
1.3.3.1 酸水解法
将78g[III]加到反应瓶中,另取84mL浓硫酸和115mL水(慢慢把酸加到搅动的水中)配成硫酸水溶液,冷至室温后再加到反应瓶中,开始搅拌,加热回流3~5h,取样分析检测反应进程,待反应完全后,冷至室温,然后将反应溶液边搅拌边倒入200mL冷水中,要使析出的固化物分散不结块,放置片刻,待固化物硬化,过滤,用少量冷水洗涤,干燥,减压(147℃/12mm或113℃/1.0mm)蒸馏得[IV]68g,收率76%,m.p.29~30℃,b.p.264~265℃
1.3.3.2 碱水解法
取84g(0.63mol)[III]和浓度15%~20%氢氧化钠水溶液约200~250mL加到反应瓶中,搅拌加热回流8h,同时在冷凝管上口排出氨气,在水解期间,若碱液不足可适当补加,至水解反应完全(无氨气排出),冷至室温,过滤反应液,得清澈红色滤液,在搅拌下加盐酸酸化,收集产物[IV],用少量冷水洗涤,干燥,减压(147℃/12mm或113℃/1.0mm)蒸馏得产物[IV]73g,收率76%。
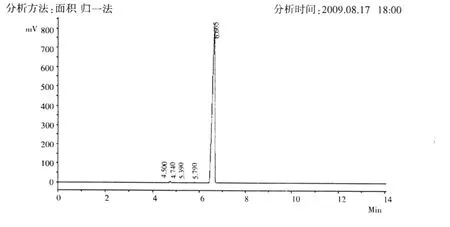

序号123456峰号123456组份名 保留时间4.500 4.740 5.390 5.790 6.665 7.415峰高394.2 36099.7 756.0 1720.5 817589.8 1328.7峰面积669.9 70275.0 1733.6 3816.1 6146512.2 2242.3含量0.0108 1.1289 0.0278 0.0613 98.7352 0.0360合计: 857888.9 6225249.1 100.0000
2 结果与讨论
(1)合成路线特点是:所用各种化工原料易得,价廉,中间产物[II]和[III]也是手性的,可以作为它用,本工艺技术易于工业化。
(2)氯化氢加成苯乙烯[I]反应,Moye[6]的实验结果:除加成物[II]外,还有[I]的二聚物。若直接蒸馏其混合物,二聚物要占15%,而采用高效分馏柱进行精馏,在恒温下得95%的馏分,二聚物占5%。此结果给了我们启示:蒸馏[II]若局部温度过高或过快,[II]易发生逆反应,脱去氯化氢,[I]的二聚物增多。同时,我们对气体在体系中的分布,气泡大小,以及进气速度等都进行优化,使设备满足工艺要求,取得了好结果。
(3)用工业产的30%氰化钠水溶液使卤代物氰化既经济、操作又方便,是常用方法。水相和有机相反应,溶剂的选择较重要,我们试用了多种溶剂。氰化钠水溶液碱性较强,所以要控制好体系的pH值,一般在弱酸性时对反应有利,蒸馏脱溶剂时,也蒸馏出部分水,分离出水的量减少,同时为弱酸性,几乎无含氰废水。
(4)水解氰化物可采用酸或碱二种方式,要比较其优缺点,一定要考虑到底物和产物对酸、碱的稳定性,所以我们采取了两种方式水解氰化物,以作比较。
[1]Robert Roger,et al.Derivatives of the Hydratropic Acids and Hydratropic Alcohols[J].Journal of Chemical Society,1960:627-629.
[2]Cynthin A Challener.手性中间体手册[M].北京:化学工业出版社,2004:476-491,513-586.
[3]Ernest L.Eliel,et al.Reduction of 2-Chloro-2-phenylpropionic Acid[J].The Journal of American Chemical Society,1952,74(4):923-928.
[4]William A.Bonner.Stereochemical Paths of Reductive Desulfuration[J].The Journal of American Chemical Society,1952,74(4):1034-1039.
[5]Isaac Angres,et al,Configuration Determination of(R)-(+)-1,1,2-Triphenylpropane[J].Journal of Organic Chemistry,1975,40(10):1957-1460.
[6]Moye C J.The Purity of α-Chloroethylbenzene[J].Australian Journal of Chemistry,1967,(20):779-782.
