基于关联分析挖掘小麦SDS 沉降值相关候选基因及KASP 标记开发
2024-12-31刘海霞张寅辉庄蕾郭梦娇赵李吴美娟侯健李甜刘红霞张学勇郝晨阳






摘要:SDS沉降值是小麦品质育种的一项重要指标。对145份小麦品种的SDS沉降值进行了全基因组关联分析,在染色体3DL 末端鉴定到成簇的显著信号,结合peak SNP 的位置与LD 衰减距离锁定587.514~589.514 Mb为候选区段;利用重测序数据(http://wheat.cau.edu.cn/WheatUnion/)发掘该区段内候选基因的变异位点,再对SDS沉降值的候选基因进行关联分析。综合关联结果、基因注释和基因表达分析,获得沉降值相关候选基因TraesCS3D03G1092400,命名为TaAGAP-3D。基于该基因的多态性SNP位点开发了可供育种利用的KASP标记Sv-3D-KASP,发现TaAGAP-3D(C)为SDS沉降值相关的优异等位变异。研究结果将为小麦SDS沉降值的遗传改良提供有效分子标记,同时也为进一步克隆品质基因提供参考。
关键词:小麦;SDS沉降值;全基因组关联分析;候选基因关联分析;KASP标记doi:10.13304/j.nykjdb.2024.0895
中图分类号:S511 文献标志码:A 文章编号:10080864(2024)12001812
小麦(Triticum aestivum L.)是世界上重要的粮食作物之一,在全球各地广泛种植,以小麦为主要口粮的人口占世界总人口的35%~40%[1]。随着生活水平的提高和消费结构的转变,人们更趋向于营养、丰富和健康的饮食消费。因此,培育优质专用小麦已成为现阶段小麦育种的重要目标。十二烷基硫酸钠(sodium dodecyl sulfate,SDS)沉降值是小麦品质育种的一项重要指标[2],其与蛋白质含量、湿面筋含量和稳定时间等呈显著正相关[3],同时SDS沉降值是由多基因控制的数量性状,具有较高的遗传力,能够用于育种早代选择[45]。
关联分析(association analysis),又称为关联作图(association mapping)或连锁不平衡作图(linkagedisequilibrium mapping),其基于基因位点间的连锁不平衡(linkage disequilibrium,LD)进行群体内基因型与表型的相关性分析,以此来确定与目标性状密切相关的基因位点或标记位点[6]。根据基因型鉴定范围的不同可将关联分析分为全基因组关联分析(genome-wide association study,GWAS)和候选基因关联分析(candidate gene association study,CGAS)[7]。
随着高通量测序技术的发展以及模型的更新,GWAS已成为解析作物复杂数量性状的有力工具。席甜甜等[8]通过对337份小麦品种籽粒相关性状进行GWAS,定位到49个稳定的显著单核苷酸多态性(single nucleotide polymorphism,SNP)位点。董一帆等[9]利用90K SNP芯片对259份小麦品种的沉降值、蛋白质含量等品质性状进行GWAS,检测到44个显著关联位点。CGAS是进行候选基因挖掘和功能验证的一种有效手段,可以借助前人的QTL作图、表达谱等研究结果,优先选择与目标性状相关的候选基因,结合表型数据、群体结构等进行候选基因关联分析[10]。Ehrenreich等[11]基于275份拟南芥材料对51个开花时间相关基因进行关联分析,进一步鉴定到2~10个与开花时间显著相关的基因,其中CO 基因是最可能的候选基因。Wilson等[12]对玉米籽粒淀粉含量相关的6个候选基因进行了关联分析,发现这些基因分别影响籽粒组分性状、淀粉糊化特性和直链淀粉水平。上述结果表明,CGAS是候选基因挖掘及基因功能验证的重要方法。
本研究以145份小麦品种为材料,结合重测序数据和多年多点SDS沉降值表型数据进行全基因组关联分析,挖掘相关候选区段,进一步通过候选基因关联分析筛选目标基因,根据目标基因的差异位点开发竞争性等位基因特异性PCR(kompetitiveallele-specific PCR,KASP)标记,并设计衍生限制性内切酶多态性标记(derived cleaved amplifiedpolymorphic sequences,dCAPS)加以验证,为小麦品质基因克隆及分子机制解析提供科学依据。
1 材料与方法
1.1 供试材料与田间试验设计
本研究以实验室构建的145份重测序自然群体为材料,包含100份育成品种,25份地方品种和20份国外引进品种[13]。试验材料分别种植在中国农业科学院作物科学研究所新乡试验基地(113°54′ E,35°18′ N)和中国农业科学院作物科学研究所顺义试验基地(116°65′ E,40°13′ N)。每份材料种植4行,行长2 m,行距25 cm,人工点播,每行播种40粒。田间管理依据当地农业生产实际进行管理,材料成熟后选取中间两行全株收获并脱粒晒干。
本研究试验材料的鉴定环境包括2018/2019年河南新乡(E1)、2019/2020 年河南新乡(E2)、2019/2020年北京顺义(E3)、2020/2021年河南新乡(E4)、2020/2021 年北京顺义(E5)、2021/2022年河南新乡(E6)、2021/2022 年北京顺义(E7)以及上述7个环境的最佳线性无偏估计(best linearunbiased prediction,BLUP)。
1.2 微量SDS 沉降值测定
将待试样品按GB5497—1985[14]定温定时法测定面粉含水量。用80 目筛旋风磨(型号Cyclotec 1093,FOSS公司,德国)将小麦磨成全粉,参照马传喜等[5]方法测定微量SDS沉降值。具体步骤如下:称取全麦粉2 g,装入35 mL的专用带塞试管中,加入16.7 mL溴酚蓝溶液,充分混匀后放置到沉淀值测定仪(型号Y15,YUCEBAS公司,土耳其)上摇5 min;取下试管再加入16.7 mLSDS-乳酸混合液(称取20 g SDS,加入20 mL乳酸储备液,用水溶解并定容至1 L),充分摇匀,放置到测定仪上摇5 min;取下试管竖立静止放置5 min,记录试管中沉淀体积。
每个样品2次重复,取其平均值。以面粉含水量14%(质量分数)湿基计算沉降值(mL),公式如下。
1.3 表型数据统计
采用IBM SPSS Statistics 27.0 软件(https://www.ibm.com/spss)和Microsoft Excel 2021对表型数据进行t 检验、方差分析和基本描述性统计。利用R语言“lme4”程序包[15]计算BLUP值,以基因型为固定效应、环境为随机效应,计算遗传力(公式2)。通过R语言绘制不同环境下SDS沉降值的频率分布图和相关性分析图。
1.4 全基因组关联分析
基因型数据来自实验室前期获得的重测序数据[13],本研究利用VCFtools-1.15对其进行质控,共鉴定到37 466 916个高质量的多态性SNP位点。质控参数为--minGQ 8 --minDP 12 --max-missing 0.9 --maf0.05 --min-alleles 2 --max-alleles 2。群体结构、LD衰减距离及主成分分析等参照已发表结果[13],即群体结构和主成分均为3,LD衰减距离为2 Mb。
全基因组关联分析采用GEMMA(genomewideefficient mixed-model association)软件的混合线性模型(mix linear model,MLM)[16],利用Q+K矩阵矫正群体结构和亲缘关系的影响。为了进一步控制假阳性,显著性阈值设定为Plt;1.0×10-6。
通过R语言将关联分析的曼哈顿图和Q-Q图进行可视化。通过LDBlockShow v1.39 软件[17]统计区段内变异位点的连锁不平衡程度并绘制热图。基于expVIP8(http://www. wheat-expression.com/)分析基因表达模式。
1.5 候选基因关联分析
在显著关联候选区段发掘的基础上,利用实验室前期的重测序数据[13](http://wheat.cau.edu.cn/WheatUnion/)查询区段内所有候选基因的序列变异(包含上下游及3’/5’-UTR区的变异),提取全部变异位点作为基因型数据。候选基因关联分析使用GAPIT v3.0包(genomic association and predictionintegrated tool version 3.0)[18]的贝叶斯信息与连锁不平衡迭代嵌套式模型(bayesian-information andlinkage-disequilibrium iteratively nested keyway,BLINK)[19]。依据Bonferroni 方法,以Plt;1.0×10-4 为阈值判定SNP标记与目标性状显著关联。
1.6 KASP 标记开发与验证
根据候选基因显著关联位点设计特异性KASP引物(WheatOmics v1.0网站,http://wheatomics.sdau.edu.cn/),包含2条带有荧光接头的分型上游引物F1-FAM 和F2-HEX 以及1 条通用下游引物Common-primer-R,引物序列见表1。KASP引物由北京六合华大基因科技有限公司合成,反应在384孔荧光定量PCR板上进行。混合反应体系5 μL∶2.2 μL DNA(20~40 ng·μL-1),2.5 μL KASP Mastermix 和0.3 μL Primer mix(F1-FAM、F2-HEX、Common-primer-R分别3.0、3.0、7.5 μL,利用ddH2O加到25 μL)。PCR扩增程序:95 ℃预变性10 min;95 ℃变性20 s,62 ℃退火40 s,10个循环;95 ℃变性20 s,58 ℃退火40 s,37个循环;28 ℃读板 1 min。采用QuantStudioTM 7 Flex 荧光定量仪(appliedbiosystems by life technologies)对扩增产物进行荧光信号检测,QuantStudioTM Real-time PCR Software v1.3(applied biosystems by life technologies)软件实现数据的可视化及结果判读。通过OriginPro 2024软件(https://www.originlab.com/)对KASP 变异与SDS 沉降值的箱线图进行可视化。
1.7 dCAPS 扩增与酶切
为了验证KASP标记的有效性,进一步利用dCAPS Finder 2.0网站(http://helix.wustl.edu/dcaps/dcaps.html)设计特异性dCAPS引物,包含一轮特异性正向引物dCAPS-F1 和反向引物dCAPS-R1(表1),该引物扩增包含变异位点的585 bp PCR产物。第2次PCR时,在SNP位点上游引入GA变异,包含特异性正向和反向引物dCAPS-F2 和dCAPS-R2(表1),并形成内切酶Sal Ⅰ的识别位点“GTCGAC”。利用内切酶Sal Ⅰ对二次PCR产物进行酶切,酶切产物在4%的琼脂糖凝胶中电泳。
具体的实验步骤如下:首先进行一轮PCR,反应体系为DNA(50 ng·μL-1) 2 μL,2×Mix 10 μL,dCAPS-F1 (10 μmol·L-1) 1 μL, dCAPS-R1(10 μmol·L-1) 1 μL,加ddH2O补至20 μL。PCR扩增程序如下:95 ℃变性3 min;95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸30 s,35 个循环;72 ℃延伸10 min,12 ℃保温。然后,将一轮PCR 产物稀释200倍作为二轮PCR的模板,引物为dCAPS-F2和dCAPS-R2,使用2×Mix进行二轮PCR,体系及扩增程序同上。利用内切酶Sal Ⅰ对二轮PCR产物进行酶切孵育,孵育条件为37 ℃,4 h。反应体系为:PCR 产物5 μL,10×Buffer 1 μL,限制性内切酶0.2 μL,ddH2O补至10 μL。最后,酶切产物在4%的琼脂糖凝胶电泳(120 V稳压电泳20 min)。
2 结果与分析
2.1 SDS 沉降值表型变异分析
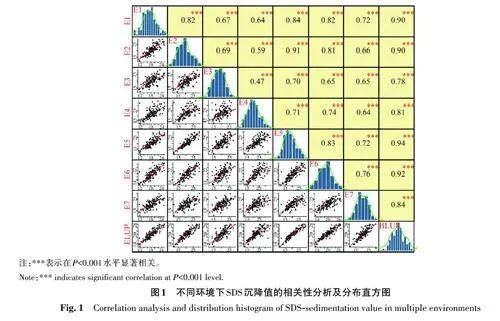
145份供试材料多年多点的SDS沉降值基本描述性统计分析结果如表2所示。可以看出,不同环境下SDS 沉降值的变异系数在14.15%~19.30%,其广义遗传力为94.10%,这表明SDS 沉降值主要受遗传因素影响,供试材料间存在较丰富的遗传变异。SDS沉降值的偏度和峰度绝对值均小于1,且频率分布直方图均呈现出正态分布(图1),表明SDS沉降值是由多基因控制的数量性状,适用于后续的关联分析。相关性分析(图1)发现,环境间的SDS沉降值呈极显著正相关,相关系数在0.47~0.94(Plt;0.001)。
2.2 SDS 沉降值全基因组关联分析
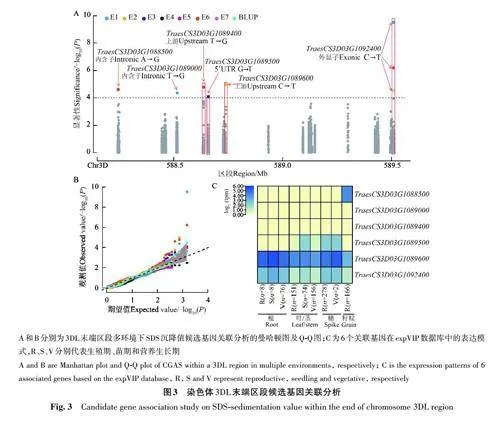
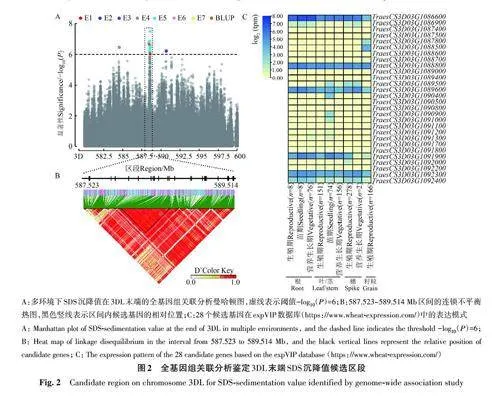
基于质控后的重测序基因型数据,利用GEMMA软件的MLM模型对多环境下的SDS沉降值进行全基因组关联分析。根据LD衰减距离,将peak SNP位置前后1 Mb设定为候选区段。在3DL末端发现成簇的显著信号(Plt;1.0×10-6),参照peakSNP的物理位置,锁定587.514~589.514 Mb为候选区段(图2A)。该区段内变异位点的连锁不平衡分析发现(图2B),多个LD区域与显著位点强连锁,进一步表明该区段与SDS沉降值存在较强的关联性。基于WheatOmics v1.0 网站(http://wheatomics.sdau.edu.cn/)查询发现,该区段包含28个高置信基因,其中通过expVIP 数据库(https://www.wheat-expression.com/)预测显示9个基因在籽粒中表达(图2C)。
2.3 3DL 末端区段候选基因关联分析
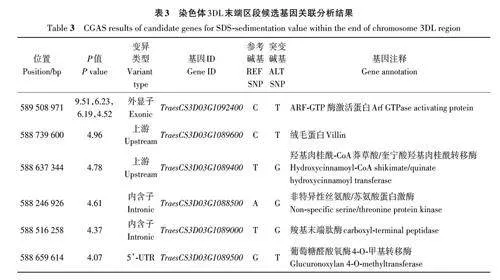
利用重测序数据(http://wheat. cau. edu. cn/WheatUnion/)查询28个高置信基因的变异位点,其中21个基因存在变异位点,获得1 473个SNPs。结合多年多点的SDS沉降值表型数据,利用GAPITv3.0包的BLINK模型进行候选基因关联分析,结果(图3A、B)表明,6 个SNP 位点显著关联(Plt;1.0×10-4),其中1个位点在4个环境下均显著关联(图3A、表3)。进一步对关联位点进行注释,4个位点为启动子上游,5’-UTR和编码区变异,而其他2个位点为内含子变异(图3A,表3)。有趣的是,这6个关联位点分别位于6个基因上,这些基因的表达量分析发现仅有3个基因在籽粒中表达。结合表达量分析和变异位点注释,初步锁定TraesCS3D03G10-92400 和TraesCS3D03G1089600 为候选基因。基因注释表明TraesCS3D03G1092400 编码GTP相关的激活蛋白(Arf GTPase activating protein),而TraesCS3D0-3G1089600 编码绒毛蛋白(Villin)。综合上述结果,推测TraesCS3D03G1092400 为SDS沉降值的候选基因,并将其命名为TaAGAP-3D。
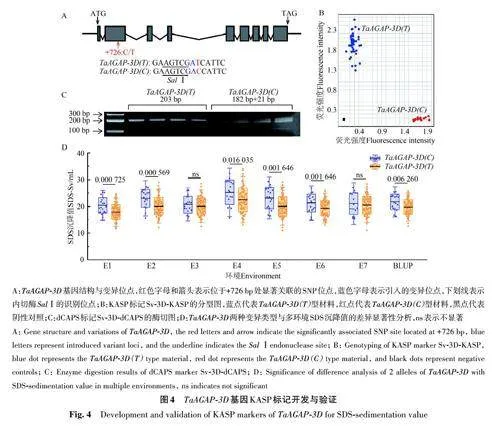
2.4 TaAGAP-3D 基因KASP 标记开发与验证
根据TaAGAP-3D 编码区上SNP 的两种变异类型(C/T)(图A),开发了有效区分它们的KASP标记Sv-3D-KASP(图B)。为了进一步验证KASP标记的准确性,同时设计了dCAPS 标记Sv-3DdCAPS-1(图4C),该标记的分型结果与KASP 标记基本吻合(表4)。进一步结合多年多点的SDS沉降值数据验证KASP标记的遗传效应,发现在大多数环境下TaAGAP-3D(C)型材料的SDS 沉降值显著高于TaAGAP-3D(T)型材料(图4D),表明TaAGAP-3D(C) 是SDS 沉降值相关的优异等位变异。
3 讨论
3.1 小麦3D 染色体上沉降值关联区段的多效性分析
小麦是异源六倍体作物,与A、B亚基因组相比,其D亚基因组的遗传多样性严重匮乏,是小麦品种改良的重要瓶颈[2021]。本研究在3D染色体的末端定位到1个SDS沉降值候选区段,其物理位置为587.523~589.514 Mb。陈华斌[22] 利用90KSNP芯片鉴定272份小麦材料,结合蛋白质含量进行了全基因组关联分析,发现在3DL末端检测到多个显著位点(IWB18421、IWB42714、IWB6798、IWB2250 和IWB58810),其物理位置相近,分别为596.089、596.718、596.916、596.916 和609.626 Mb;基于全基因组LD衰减距离18.3 Mb,该研究将这些位点划分到了同一QTL区段,而该区段涵盖了本研究鉴定到的位点。彭小爱等[23]对118份小麦材料的55K SNP芯片基因型数据和容重进行全基因组关联分析,在3个环境下均定位到了3D染色体上的显著关联位点AX-108887454,其物理位置为590.04 Mb;基于作者报道的8 Mb全基因组LD衰减距离,该候选区段同样包含了本研究定位到的区段。这些结果表明本研究鉴定的3DL末端区段可能控制多个品质性状,其遗传基础有待进一步深入解析。
3.2 联合多种关联分析缩小候选区段及预测候选基因
全基因组关联分析(GWAS)是对全基因组范围内的基因型与表型进行关联分析的方法,根据成簇的显著信号及LD衰减距离定位候选区段[24]。而候选基因关联分析(CGAS)则通过目标区段内候选基因的变异位点与表型展开关联分析,利用统计分析挖掘与目标性状相关的基因或功能位点[25],从而进一步筛选候选基因。前人的研究大多数仅考虑使用GWAS或CGAS单一方法鉴定候选区段或基因,如Wen 等[26]利用重测序技术对121份棉花种质资源进行GWAS,检测到与株高和果枝数量相关的11 个QTLs;Setter 等[27] 对玉米540个基因的1 229个SNP进行候选基因关联分析,发现一些基因的SNP与糖和脱落酸相关。前人也联合GWAS与其他组学分析进行共定位以缩小候选区段和预测候选基因,如Tang 等[28]结合GWAS和转录组分析来解析505份甘蓝型油菜含油量的遗传基础,通过GWAS鉴定到27个与含油量显著相关的QTLs后,进一步利用转录组分析锁定了候选基因;Anacleto等[29]联合GWAS和转录组关联分析(transcriptome-wide association study,TWAS)对305 份籼稻的血糖生成指数(glycemicindex,GI)进行分析,GWAS在 6号染色体上鉴定到1 个与GI 性状相关且包含26 个基因的区段,TWAS分析进一步将该区段内的候选基因缩小至13个。本研究通过联合GWAS和CGAS有效缩小了与SDS沉降值相关的候选区段并预测了候选基因(图2A和3A)。因此,全基因组关联分析联合其他多种分析方法如候选基因关联分析,可以更有效地发掘作物重要农艺性状基因。
本研究预测的候选基因TraesCS3D03G1092400 被注释为ARF-GTP酶激活蛋白,关联分析表明该基因与SDS沉降值相关。鉴于水稻中已克隆了1个具有相同注释的基因OsAGAP[30],因此将其命名为TaAGAP-3D。研究表明,OsAGAP 能够调节生长素内流、囊泡运输和根发育[31];并且在小麦中发现GPC-B1 编码NAC转录因子(NAM-B1),RNA干扰后延迟衰老并使小麦籽粒的蛋白质、锌和铁含量降低30%以上[32]。综上所述,推测TaAGAP-3D可能响应生长素调节信号使营养物质进行再分配,其基因功能和分子机制有待通过多种分子生物学方法进一步研究。
3.3 小麦品质相关KASP 标记开发与应用
随着分子生物技术的快速发展,分子标记辅助选择已经成为现代分子育种的有效工具[33]。基于SNP的KASP基因分型技术具有灵活性强、高通量、成本低及操作简单高效等特点,在分子标记辅助选择育种中得到广泛的应用。在小麦中,KASP技术多用于抗病和耐非生物胁迫相关基因的研究[34],而在品质方面的分子标记还较少。魏广源等[35]通过硒含量相关的SNP位点开发KASP标记AX-1和AX-2,该标记能够有效区分高硒和低硒籽粒样品并用于高硒小麦选育。Liu等[36]基于Gli-γ1-1D 的2种主要单倍型(Gli-γ1-Ⅰ和Gli-γ1-Ⅱ)开发了KASP标记,206份小麦材料基因型鉴定发现携带Gli-γ1-Ⅰ材料的SDS沉降值显著高于携带Gli-γ1-Ⅱ材料,这为Gli-γ1-Ⅰ在小麦品质分子育种中的利用提供了可靠标记。毕俊鸽等[37]针对QTL定位到的与籽粒蛋白质含量相关的候选基因开发了2个KASP标记并在166份国内外小麦材料中进行验证,用于筛选高籽粒蛋白质含量的优异资源。本研究基于TaAGAP-3D 多态性SNP 位点开发了KASP 标记Sv-3D-KASP,并发现TaAGAP-3D(C)为SDS沉降值相关的优异变异,将为小麦品质分子标记辅助育种提供新基因和新标记。
参 考 文 献
[1] LIU Y X, LIN Y, GAO S, et al .. A genome-wide association study of 23 agronomic traits in Chinese wheat landraces [J].Plant J., 2017,91(5):861-873.
[2] 范金平,吕国锋,张伯桥,等.SDS沉降值在小麦良种繁育中的应用[J].安徽农业科学, 2003(6):1043-1044.
FAN J P, LYU G F, ZHANG B Q, et al .. Application of SDS sedimentation value in wheat breeding [J]. J. Anhui Agric. Sci.,2003(6):1043-1044.
[3] 王峰.小麦品质性状的遗传变异和诱变研究[D].合肥:安徽农业大学,2013.
WANG F. Studies on genetic variation and mutation of quality traits in wheat [D]. Hefei: Anhui Agricultural University, 2013.
[4] 夏云祥,陈辉,司红起,等. 小麦微核心种质中育成品种和地方品种的SDS沉降值分析[J]. 粮食与饲料工业,2011(10):12-14.
XIA Y X, CHEN H, SI H Q, et al .. Analysis of SDSsedimentation volume in bred and local varieties of Chinese wheat micro-core collections [J]. Cereal Feed. Ind., 2011(10):12-14.
[5] 马传喜,徐风,汪心乐.影响SDS沉降值的试验因素分析[J].安徽农业大学学报,1995, 22 (1):1-6.
MA C X, XU F. WANG X L. Analysis of experimental factors affecting SDS sedimentation value [J]. J. Anhui Agric. Univ.,1995,22(1) :1-6.
[6] FLINT-GARCIA S A, THORNSBERRY J M, BUCKLER E S. Structure of linkage disequilibrium in plants [J]. Annu. Rev.Plant Biol., 2003,54:357-374.
[7] ZHU C S, GORE M, BUCKLER E S, et al .. Status and prospects of association mapping in plants [J]. Plant Genome,2008, 1(1): 5-20.
[8] 席甜甜,吴倩,杨建光,等. 337份小麦品种籽粒相关性状的全基因组关联分析[J]. 麦类作物学报,2024,44(5):547-558.
XI T T, WU Q, YANG J G, et al .. Genome-wide association studies of 337 wheat varieties for grain-related traits [J]. J.Triticeae Crops, 2024,44(5):547-558.
[9] 董一帆,任毅,程宇坤,等. 冬小麦籽粒主要品质性状的全基因组关联分析[J]. 中国农业科学,2023,56(11):2047-2063.
DONG Y F, REN Y, CHENG Y K, et al .. Genome-wide association study of grain main quality related traits in winter wheat [J]. Sci. Agric. Sin., 2023,56(11):2047-2063.
[10] 王荣焕,王天宇,黎裕.关联分析在作物种质资源分子评价中的应用[J].植物遗传资源学报,2007,8(3):366-372.
WANG R H, WANG T Y, LI Y. Application of association analysis in molecular evaluation of crop germplasm resources[J]. J. Plant Genet. Resour., 2007,8(3):366-372.
[11] EHRENREICH I M, HANZAWA Y, CHOU L, et al ..Candidate gene association mapping of Arabidopsis flowering time [J]. Genetics, 2009,183(1):325-335.
[12] WILSON L M, WHITT S R, IBANEZ A M, et al . Dissection of maize kernel composition and starch production by candidate gene association [J]. Plant Cell, 2004, 16(10): 2719-2733.
[13] HAO C Y, JIAO C Z, HOU J, et al .. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China [J]. Mol. Plant, 2020,13(12):1733-1751.
[14] 高修吾,杨浩然,吴艳霞,等.粮食、油料检验 水分测定法:GB5497—1985[S].北京:中国标准出版社,1985.
[15] BATES D, MACHLER M, BOLKER B, et al .. Fitting linear mixed-effects models using lme4 [J]. J. Stat. Software, 2015,67(1):1-48.
[16] ZHOU X, STEPHENS M. Genome-wide efficient mixed-model analysis for association studies [J]. Nat. Genet., 2012, 44(7):821-824.
[17] DONG S S, HE W M, JI J J, et al .. LDBlockShow:a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files [J/OL].Brief. Bioinform., 2021, 22(4): bbaa227 [2024-11-05]. https://doi.org/ 10.1093/bib/bbaa227.
[18] WANG J B, ZHANG Z W. GAPIT version 3:boosting power and accuracy for genomic association and prediction [J].Genom. Proteom. Bioinform., 2021,19(4):629-640.
[19] HUANG M, LIU X L, ZHOU Y, et al .. BLINK:a package for the next level of genome-wide association studies with both individuals and markers in the millions [J]. Gigascience, 2019,8(2):giy154 [2024-11-06]. https://doi.org/10.1093/gigascience/giy154.
[20] VOSS-FELS K, FRISCH M, QIAN L W,et al .. Subgenomic diversity patterns caused by directional selection in bread wheat gene pools [J/OL]. Plant Genome, 2015, 8(2): 03.0013[2024-11-06]. https://doi.org/10.3835/plantgenome2015.03.0013.
[21] PONT C, LEROY T, SEIDEL M, et al .. Tracing the ancestry of modern bread wheats [J]. Nat. Genet., 2019,51(5):905-911.
[22] 陈华斌.小麦粒形、品质和农艺相关性状的全基因组关联分析[D].杭州:浙江大学, 2021.
CHEN H B. Genome-wide association study of grain shape, quality and agronomic related traits in wheat [D]. Hangzhou:Zhejiang University, 2021.
[23] 彭小爱,卢茂昂,张玲,等.基于55K SNP芯片的小麦籽粒主要品质性状的全基因组关联分析[J].作物学报, 2024, 50(8):1948-1960.
PENG X A, LU M A, ZHANG L, et al .. Genome-wide association study of major grain quality traits in wheat based on 55K SNP arrays [J]. Acta Agron. Sin., 2024, 50(8):1948-1960.
[24] 任生林,吴才文,经艳芬,等.全基因组关联分析在作物中的研究进展[J].分子植物育种,2024,22(11):3594-3602.
REN S L, WU C W, JING Y F, et al .. Research progress of genome-wide association analysis in crops [J]. Mol. Plant Breeding, 2024, 22(11): 3594-3602.
[25] 吴宁宁.玉米种质粒型及粒重性状的鉴定和全基因组关联分析(GWAS)[D].杭州: 浙江农林大学, 2023.
WU N N. Identification of grain type and grain weight traits in maize germplasms and genome-wide association analysis (GWAS) [D]. Hangzhou: Zhejiang Aamp;F University, 2023.
[26] WEN T W, DAI B S, WANG T, et al .. Genetic variations in plant architecture traits in cotton (Gossypium hirsutum)revealed by a genome-wide association study [J]. Crop J., 2019,7(2):209-216.
[27] SETTER T L, YAN J B, WARBURTON M, et al .. Genetic association mapping identifies single nucleotide polymorphisms in genes that affect abscisic acid levels in maize floral tissues during drought [J]. J. Exp. Bot., 2011,62(2):701-716.
[28] TANG S, ZHAO H, LU S P, et al .. Genome- and transcriptomewide association studies provide insights into the genetic basis of natural variation of seed oil content in Brassica napus [J]. Mol. Plant, 2021,14(3):470-487.
[29] ANACLETO R, BADONI S, PARWEEN S,et al .. Integrating a genome-wide association study with a large-scale transcriptome analysis to predict genetic regions influencing the glycaemic index and texture in rice [J]. Plant Biotechnol. J., 2019,17(7):1261-1275.
[30] ZHUANG X L, XU Y, CHONG K, et al .. OsAGAP,an ARFGAP from rice,regulates root development mediated by auxin in Arabidopsis [J]. Plant Cell Environ., 2005,28(2):147-156.
[31] ZHUANG X L, JIANG J F, LI J H, et al.. Over-expression of OsAGAP, an ARF-GAP, interferes with auxin influx, vesicle trafficking and root development [J]. Plant J., 2006,48(4):581-591.
[32] UAUY C, DISTELFELD A, FAHIMA T, et al .. A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat [J]. Science, 2006,314(5803):1298-1301.
[33] COLLARD B C, MACKILL D J. Marker-assisted selection:an approach for precision plant breeding in the twenty-first century [J]. Philos. Trans. R. Soc. Lond. Ser. Biol. Sci., 2008, 363(1491):557-572.
[34] 杨青青,唐家琪,张昌泉,等. KASP标记技术在主要农作物中的应用及展望[J]. 生物技术通报,2022,38(4):58-71.
YANG Q Q, TANG J Q, ZHANG C Q, et al .. Application and prospect of KASP marker technology in main crops [J].Biotechnol. Bull., 2022,38(4):58-71.
[35] 魏广辉,李执,陈强,等. 人工合成小麦SHW-L1 高硒含量KASP 分子标记开发及其应用[J]. 中国农业科学,2020,53(20):4103-4112.
WEI G H,LI Z,CHEN Q,et al .. Development and utilization of KASP marker for Se concentration in synthetic wheat SHW-L1[J]. Sci. Agric. Sin., 2020,53(20):4103-4112.
[36] LIU D, YANG H, ZHANG Z, et al .. An elite γ-gliadin allele improves end-use quality in wheat [J]. New Phytol., 2023,239(1):87-101.
[37] 毕俊鸽,曾占奎,李琼,等. 两个RIL群体中小麦籽粒品质相关性状QTL 定位及KASP 标记开发[J]. 作物学报, 2024,50(7): 1669-1683.
BI J G, ZENG Z K, LI Q, et al .. QTL mapping and KASP marker development of grain quality-relating traits in two wheat RIL populations [J]. Acta Agron. Sin., 2024, 50(7):1669-1683.
