一株柴油降解菌Bacillus S3-2-LX的筛选鉴定和基因组测序分析
2024-11-11叶春宏杨蕊毓彭超路璐






摘 要:柴油污染对环境生物多样性和人类健康造成了严重威胁,微生物修复是目前柴油污染场地修复中的重要手段之一。以柴油为唯一的碳源,从四川某高校一垃圾堆放点的土壤中分离出一株芽孢杆菌属Bacillus的菌株S3-2-LX,菌体呈杆状,菌落呈乳白色圆形。为了验证该菌株是否具有多环芳烃降解能力,开展了多环芳烃降解培养实验,并基于Illumina HiSeq 2000平台进行de novo测序。结果表明:培养7 d后,S3-2-LX菌株对萘、菲和芘的降解率分别为99.1%、42.1%和29.2%。S3-2-LX基因组测序总长度5 869 851 bp,共得到53个Scaffold,GC含量为34.89%,能注释到COG、KEGG的基因数目分别为4211、3060个;S3-2-LX具有与烃类代谢相关的代谢通路,拥有降解烷烃、二恶英、二甲苯、苯甲酸、多环芳烃等的能力。通过对S3-2-LX的生理特性和代谢途径分析,进一步揭示了其降解柴油的关键步骤和机制。
关键词:柴油降解菌;分离和鉴定;芽孢杆菌属;基因组测序;基因注释
中图分类号:Q93-3;Q781 文献标志码:A 文章编号:1673-5072(2024)06-0569-10
石油产品在大规模生产、运输和使用中产生的污染物成为环境中普遍存在的污染[1],这些石油污染物在环境中会长期滞留,对生态环境安全构成潜在的威胁[2]。柴油作为商业石油产品的一种,是环境中常见的石油污染物,也是环境治理中的重要修复对象[3]。相较于物理和化学修复,生物修复具有更多优势,可以利用微生物的多种代谢能力去除环境中的柴油污染物,且不易导致土壤的二次污染[4]。然而,不同的土壤环境条件和柴油污染物组成可能使得施入的柴油降解微生物菌剂在污染场地环境中适应能力不足,从而限制了它们的生长速度和修复效率,影响生物修复的效果[5]。因此,研究原位土壤中的土著柴油降解微生物的生态和生理代谢潜力对实施生物修复至关重要[6]。
自然界中存在着丰富多样的柴油降解微生物,如在油田土壤[7]、海洋沉积物[8]和红树林沉积物[9]等不同环境中均能分离出具有柴油降解能力的微生物。然而,柴油污染物是一种复杂的混合物,主要由烷烃类化合物和芳香族化合物组成,不同类型的柴油降解菌对这些化合物的降解具有特异性和不同的效率[10]。Garrido等[11]从柴油污染场地的根际土壤中分离出的以Pseudomonas、Aquabacterium、Chryseobacterium和Sphingomonadaceae为主的细菌联盟能够利用不同类型的烷烃和多环芳烃(polycyclic aromatic hydrocarbons,PAHs)作为唯一的碳源和能源进行生长。Gran-scheuch等[12]从受柴油污染的南极土壤中分离出的PAHs高效降解细菌Sphingobium xenophagum D43FB能够很好地修复原位柴油污染物。组学技术的发展使得对柴油降解微生物的代谢功能基因更为明晰[13]。例如,参与烷烃降解过程的烷烃单加氧酶基因(AlkB)、烷烃单加氧酶基因(Alkm)和细胞色素P450基因(CYP)[14-15];参与PAHs降解过程的儿茶酚2,3-双加氧酶基因(xylE)、萘双加氧酶基因(ndoB)、吡喃双加氧酶基因(nidA)和芘双加氧酶大亚基基因[15-16]。因此,通过分析柴油降解菌株的基因组信息,可以挖掘其代谢柴油组分的潜力,能定向地为柴油污染场地筛选高效降解菌种进行生物修复。
因此,本研究针对一处垃圾堆放点的土壤,以柴油为添加碳源,筛选能降解柴油的微生物。同时,对分离的菌株开展了培养实验,验证其对PAHs的降解能力,并结合基因组测序技术对其代谢途径和潜力进行分析,以揭示其对柴油组分的降解潜力,为未来柴油污染场地的生物修复提供有效的菌株信息。
1 材料和方法
1.1 土壤来源
土壤样品采自四川省南充市西华师范大学校内的一厨余垃圾堆放点(106°07′08″E,30°81′ 21″N),通过无菌铲从表层(0~10 cm)土壤中采集,并将其装入塑封袋中,在-20 ℃冰箱中保存。
1.2 培养基
LB(Luria-Bertani)液体培养基:10 g胰蛋白胨,5 g酵母粉,10 g NaCl,加蒸馏水定容至1 L,调节pH为7.0~7.2。
LB固体培养基:在1 L的液体培养基中加入15 g琼脂粉,倒入灭菌的培养皿。
无机盐培养基(Mineral Salt Medium,MSM)[17]:5 mL PBS,3 mL MgSO4溶液,1 mL CaCl2溶液,1 mL FeCl3溶液,1 mL微量元素溶液,加蒸馏水定容至1 L,调节pH为7.0~7.2。
选择性无机盐液体培养基:在已灭菌的MSM中加入1%(按总体积计算)无菌柴油,并使用0.22 μm的滤膜进行过滤灭菌。
选择性无机盐固体培养基:向MSM中添加1.5%(按总体积计算)的琼脂粉,灭菌后倒入平板中。待平板凝固后,取100 μL柴油涂布于平板表面,制成油平板。
含PAHs的MSM:称量0.05 g萘(二环PAH)至100 mL培养瓶中,移至通风橱。向培养瓶中加入丙酮溶液定容至100 mL,从而制得500 mg·L-1萘母液。将培养瓶口用封口膜密封,避光保存。向已灭菌的100 mL的培养瓶中加入5 mL的萘母液,将其置于通风橱中约30 min。待丙酮挥发后,加入已灭菌的45 mL MSM,从而制得45 mL含50 mg·L-1萘的MSM。制备含50 mg·L-1菲(三环PAH)和50 mg·L-1芘(四环PAH)的MSM的方法与上述相同。
1.3 柴油降解菌的分离方法
取10 g土样加入装有100 mL无菌水的培养瓶中,置于30 ℃、180 r·min-1的恒温振荡箱中振荡3 h后静置30 min,收集上层液体作为土壤悬浊液。取10 mL土壤悬浊液接种至装有100 mL含1%柴油的选择性无机盐液体培养基中,在37 ℃、150 r·min-1的避光条件下培养3 d。从上一培养瓶中取5 mL培养液接种至45 mL含1%柴油的选择性无机盐液体培养基中,在相同条件下继续富集培养2~3 d。按照上述步骤重复富集培养3次。取最后一次富集培养得到的200 μL培养液均匀涂布于LB固体培养基上,在37 ℃恒温培养箱中过夜培养,可观察到不同形态的菌落。
对不同形态的菌落分别进行划线纯化,以实现单菌的分离。为了验证分离的菌株是否为柴油降解菌,将单菌落划线于涂有柴油的平板上,置于37 ℃恒温箱中培养,筛选出能够在该平板上生长的菌落。同时,将菌落加入到选择性无机盐液体培养基中,选择有明显乳化柴油现象的菌株进行了进一步的分子生物学鉴定,分析其系统发育水平。
使用由金斯瑞生物科技公司合成的16S rRNA通用引物(515F:GTGCCAGCMGCCGCGG;907R:CCGTCAATTCMTTTRAGTTT)进行PCR扩增和凝胶电泳实验,将目标条带的扩增产物送至北京六合华大基因科技有限公司测序。菌株用甘油封存于-80 ℃冰箱。从得到的测序结果中,选取其中一株柴油降解菌S3-2-LX的菌株序列,使用MEGA-X软件中的Neighbor-Joining方法构建系统发育树。
1.4 柴油降解菌形态和菌落的表征方法
使用涂布器将S3-2-LX菌液均匀涂布在LB固体培养基表面,在37 ℃条件下恒温培养过夜,对S3-2-LX菌株的菌落进行观察。将S3-2-LX菌液离心以去除上清液,使用4%甲醛对菌体细胞固定后再进行荧光染色,在荧光显微镜下观察S3-2-LX菌株的细胞形态。
1.5 多环芳烃降解实验
取出50%甘油管藏的菌液,在室温下进行冻融处理后,取20 μL菌液涂布在LB固体培养基上,于37 ℃恒温条件下培养直至出现单菌落。挑取单菌落并转移到50 mL LB液体培养基中,待培养基内菌群的光密度(OD600)约为0.2后,将全部菌液与LB液体培养基一同转移到50 mL无菌离心管中,5 000 r·min-1离心5 min后倒掉上清液。向离心管中加入MSM,并用涡旋混匀。再次以5 000 r·min-1离心2 min并倒掉上清液,重复此步骤2次后,向离心管中加入50 mL已灭菌的MSM以重悬菌体,制成菌悬液。取5 mL的S3-2-LX菌悬液分别接种至含有45 mL 50 mg·L-1萘、菲和芘的MSM的培养瓶中,置于37 ℃、100 r·min-1、初始pH为7的条件下培养。培养7 d后,测定培养瓶中各PAHs的残余量。
1.6 多环芳烃的测定方法
1.6.1 萃取培养液中残余的多环芳烃
在通风橱内,分别向已培养了7 d的含有不同PAH和菌悬液的培养瓶中加入5 mL正己烷,并将培养瓶放入超声仪中进行超声波萃取,持续30 min后,静置15 min。将培养瓶中的全部液体倒入50 mL离心管中,再次静置10 min,等待液体中的气泡消失。当培养液明显分层后,使用1 mL注射器沿离心管管壁吸取上层的萃取液,通过0.22 μm的绿色有机滤膜过滤后,将其打入安捷伦细胞瓶内,以备后续测定。
1.6.2 气相色谱仪测定条件
使用GC-2014型气相色谱仪进行GC-FID测定。进样量为2 μL,N2进样速度为50 mL·min-1,H2进样速度为40 mL·min-1,色谱柱采用SK-5(30.0 m×0.25 mm×0.25 μm)。具体的色谱条件为:采用不分流进样,进样口温度为280 ℃;初始柱温为100 ℃;检测器温度为300 ℃。柱箱采用升温程序:初始温度为100 ℃,保持1 min;以15 ℃·min-1的升温速率升温至255 ℃,保持1 min;再以1 ℃·min-1的升温速率升温至265 ℃,保持1 min;最后以2.5 ℃·min-1的升温速率升温至295 ℃,保持5 min。
1.6.3 GC-FID定量分析多环芳烃含量
本实验采用外标法进行定量测定。使用含有萘、菲和芘的混合标准溶液(稀释梯度为10.0、7.5、5.0、2.5和1.0 mg·L-1)进行GC-FID定量分析,基于不同浓度的峰面积绘制线性回归方程,对土壤样品中的PAHs进行定量。在相同的色谱条件下,依次对每个处理的样品进行GC-FID测定,根据保留时间确定每个样品中PAHs的峰面积。通过将待测样品的峰面积代入线性回归方程,计算样品中PAHs的含量,并据此计算出每个样品中多环芳烃的残余浓度。
1.7 基因组测序和组装
将菌株S3-2-LX接种于LB液体培养基中,并在37 ℃下培养12 h。使用离心方法收集菌体,并将其用无菌缓冲液清洗2~3次,用于后续的基因组测序。
将S3-2-LX菌体样品寄给美吉生物公司Illumina HiSeq 2000平台进行基因组测序。在进行DNA提取后,美吉生物公司对提取到的DNA样品进行质量检测。对质检合格的DNA样品构建插入片段约为400 bp的文库,再进行PE150(pair-end)测序。对获得序列进行基因组组装,得到多条基因组scaffold。对基因组进行注释,包括对编码序列(CDS)、tRNA、rRNA和基因组岛进行预测。对预测得到的编码基因进行COG和KEGG注释,以获取相应的功能注释信息[18]。
2 结 果
2.1 S3-2-LX菌株的分离、鉴定和表征
16S rRNA基因测序分析表明,S3-2-LX菌株隶属于芽孢杆菌属(Bacillus)(Genbank序列号MZ127404),该菌株的16S rRNA基因序列与菌株Bacillus cereus group sp.strain TLPF1的相似度最高,为91.73%(图1)。
S3-2-LX菌株的菌落直径约2 mm,为圆形、乳白色的不透明菌落,菌落表面光滑,边缘完整(图2a),该菌株的菌体细胞呈杆状,长度约为2 μm(图2b)。
2.2 S3-2-LX菌株对多环芳烃的降解
S3-2-LX菌株在相同条件下培养7 d后,对萘的降解率为99.06%,明显高于对菲(42.10%)和芘(29.20%)的降解率。
2.3 S3-2-LX菌株的基因组评估
如图3所示,不同基因拼接片段的GC含量集中在同一区域,表明该基因组未受到外源物种的污染,部分基因可能来自质粒的DNA。该基因组平均深度为183.79 X ,GC含量为34.89%。
设置17-mer为参数,逐步向右移动,得到各深度的频率,以此来评估基因组的质量,并确定是否存在质粒污染。结果如图4所示:频率曲线存在一个明显的主峰,说明该菌株的测序结果可信;此外,出现的次峰表明一部分片段可能是重复片段,或者可能是具有多个拷贝的质粒。
2.4 S3-2-LX菌株的基因组组装结果
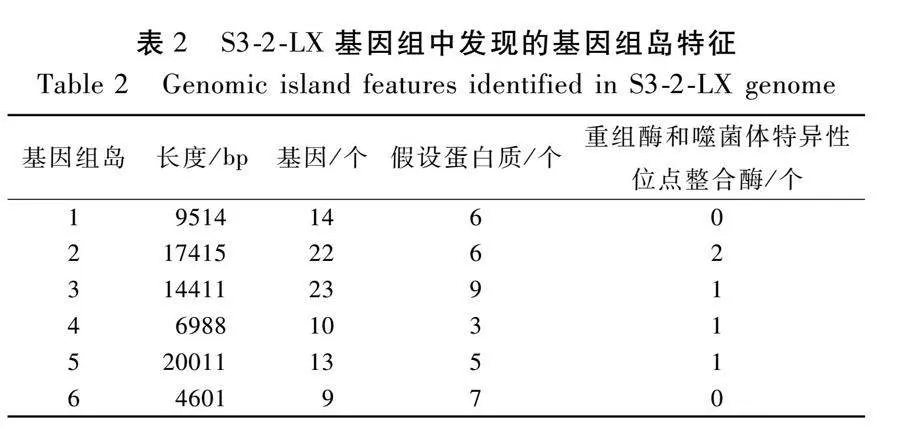
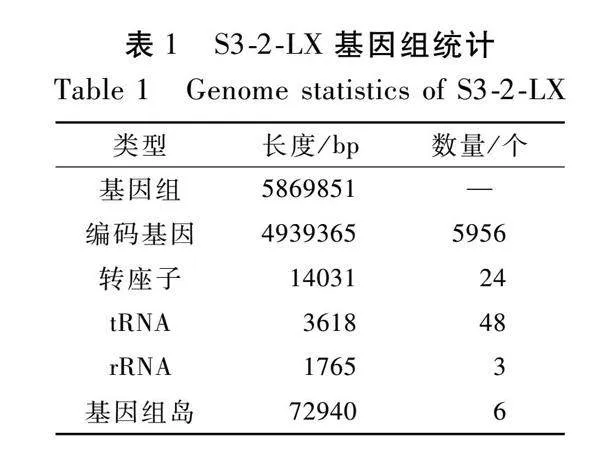
由表1可知,S3-2-LX基因组序列的总长度为5 869 851 bp。通过对S3-2-LX菌株的基因进行编码基因预测,可知其有5 956个编码基因,总长度为4 939 365 bp,占基因组的84.15%。在S3-2-LX菌株的全基因组中,共有24个转座子,总长度为14 031 bp;包含48个tRNA,总长度为3 618 bp,以及3个rRNA,总长度为1 765 bp。与多种生物功能相关的基因组区段(基因组岛,genomic island)共有6个,总长度为72 940 bp。这些基因组岛中共91个基因,其中36个基因被注释为功能未知的假设蛋白质,5个基因被注释为重组酶和噬菌体特异性位点整合酶(表2)。
2.5 S3-2-LX菌株的功能注释
2.5.1 COG数据库注释
COG对比结果显示(图5),共有4 211个基因完成了蛋白质注释,占全部基因的70.70%。其中,注释数量较多的基因功能主要包括:参与氨基酸运输和代谢(E:Amino acid transport metabolism)的基因共368个(8.74%);参与转录(K:Transcription)的基因共346个(8.22%);参与无机离子的转运与代谢(P:Inorganic ion transport and metabolism)的基因共251个(5.96%);参与碳水化合物运输和代谢(G:Carbohydrate transport and metabolism)的基因共233个(5.53%);参与细胞壁/膜/包膜生物发生(M:Cell wall/Membrane/Envelope biogenesis)的基因共228个(5.41%)。其中,参与转录的基因中包括编码典型的转录调控因子LysR型、GntR型、EamA-like型、IcIr型、MarR型和AraC型家族调节因子的基因;参与细胞运动(N:cell motility)的基因共36个(0.85%),包括编码鞭毛蛋白FliS、FlgC、FliE等的基因和编码趋化复合体蛋白CheC、CheR、CheY、CheZ的基因。
2.5.2 KEGG数据库注释
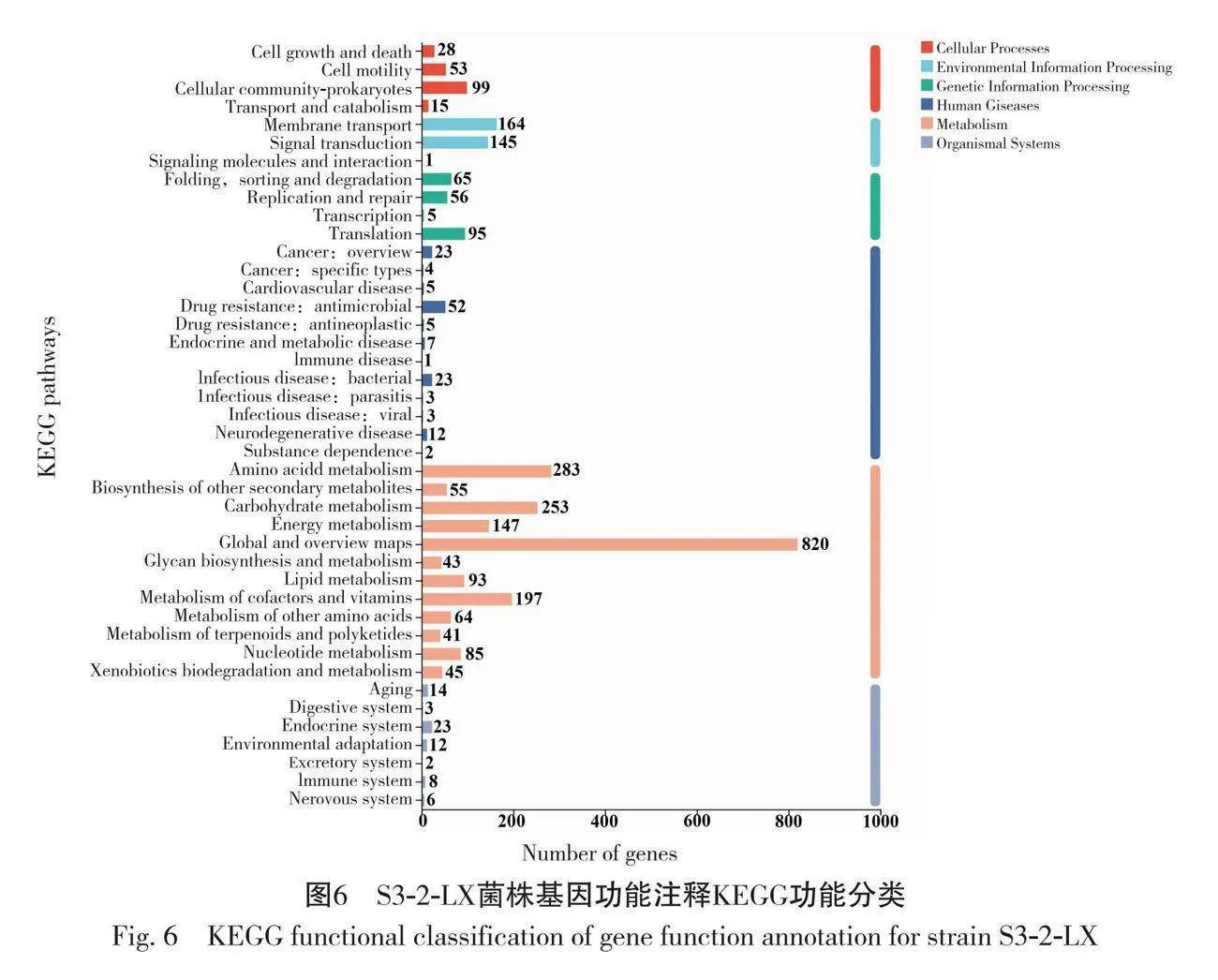
KEGG对比结果显示(图6),S3-2-LX菌株在细胞过程(Cellular Processes)、环境信息处理(Environmental Information Processing)、遗传信息处理(Genetic Information Processing)、人类疾病(Human Giseases)、新陈代谢(Metabolism)和生物体系统(Organismal Systems)这6大功能上共有3060个基因进行了注释,涵盖了42个通路。对于涉及的代谢通路总数以及每个通路参与的基因数量进行对比发现,在新陈代谢、环境信息处理、遗传信息处理和细胞过程功能上,基因的功能注释较多。富集分析显示,在代谢通路中共有2126个基因得到了注释,其中与碳水化合物代谢(Carbohydrate metabolism)相关的基因注释数量为253个,占代谢通路注释的基因总数的11.9%。涉及异种生物降解和代谢(Xenobiotics biodegradation and metabolism)的基因共有45个,占代谢通路注释的基因总数的2.12%。在环境信息处理方面共有310个基因进行了注释,在遗传信息处理理方面共有221个基因进行了注释,细胞过程中共有99个基因进行了注释,其中细胞群落-原核生物是注释基因数量最多的。此外,注释到异种生物降解和代谢的代谢通路有二恶英降解途径(ko00621)、二甲苯降解途径(ko00622)、硝基甲苯降解途径(ko00633)等(表3)。
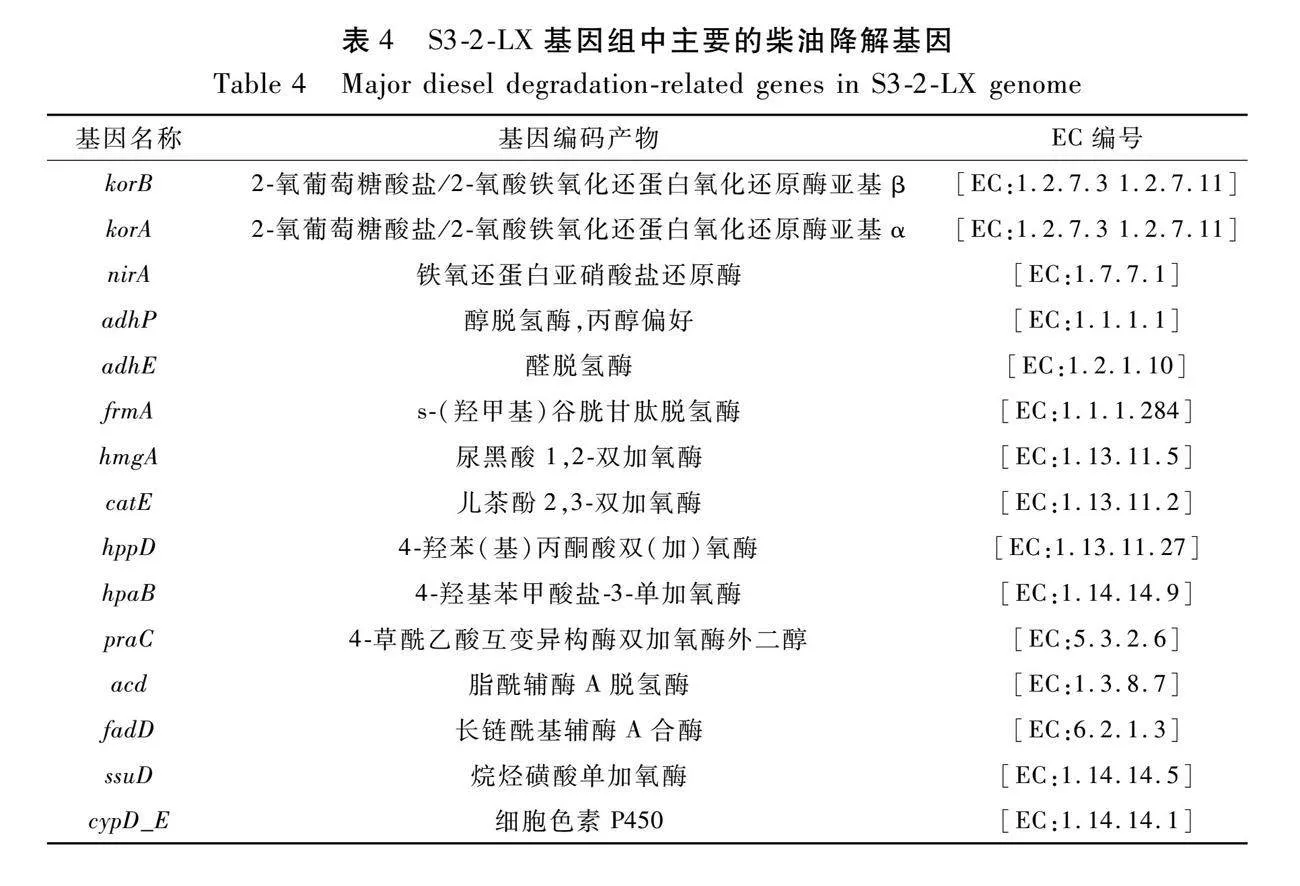
被注释的基因组主要集中在芳香族化合物降解基因上,如儿茶酚2,3-双加氧酶基因(catE)、尿黑酸1,2-双加氧酶基因(hmgA)、s-(羟甲基)谷胱甘肽脱氢酶基因(frmA)和4-草酰乙酸互变异构酶双加氧酶外二醇基因(praC)等。此外,还存在参与降解烷烃的降解酶基因,如烷烃磺酸单加氧酶基因(ssuD)、细胞色素P450基因(cypD_E)(表4)。
3 讨 论
3.1 S3-2-LX菌株的菌属特征
本研究分离出的柴油降解菌S3-2-LX菌株对于修复柴油类污染场地具有现实意义,尤其在PAHs污染较重的土壤环境中。芽孢杆菌属属于好氧或兼性厌氧的革兰氏阳性细菌,化能异养型,能够产生抵抗不利条件的特殊芽孢[19]。S3-2-LX菌株基因组中存在5个重组酶和噬菌体特异性位点整合酶的基因,该基因对水平基因转移有重要作用[20],这表明该菌株在在垃圾堆放点这一复杂多变的污染环境中具有很强的适应能力。在深海环境中分离的多环芳烃降解菌Celeribacter indicus P73T的基因组中也发现了多个转座酶基因和整合酶基因等可移动遗传元件[21]。
3.2 S3-2-LX菌株对PAHs的降解能力
S3-2-LX菌株对萘、菲和芘有较好的降解作用,且降解率随着PAHs环数的增加而降低。这一发现与以往从油田土壤、森林土壤和湿地土壤中分离的多种芽孢杆菌降解能力相似,这些菌属可以有效降解不同环数的PAHs,例如萘[22]、苯并[a]芘[23]等。Rabodonirina等[24]研究表明,在油污染的土壤中分离出的混合菌Pseudomonas和Bacillus能够有效降解芴(65%~86%)和菲(86%~95%),然而对高分子量的芘和氟蒽的降解效率则显著低于芴和菲。该规律在其他PAHs降解菌中也有所体现。例如在同一培养条件下,PAHs降解菌Bacillus spp.对萘的降解率高达89.30%,而对于其他3种较高分子量的PAHs(菲、氟蒽和芘)的降解率在45%~60%[25]。有研究表明,随着PAHs环数的增加,其疏水性和电化学稳定性也相应增加,从而导致它们在环境中的生物可利用性降低,在环境中的留存时间也随之增加。同时,降解高环PAHs的微生物大多在环境中的相对丰度较低,且生长速率较慢,这可能也是导致菲和芘的降解效率显著低于萘的原因[26]。
3.3 柴油降解菌株S3-2-LX的代谢功能基因
基因组的注释信息很好地印证了S3-2-LX菌株的PAHs降解能力。S3-2-LX菌株基因组中含有二恶英、二甲苯和萘等降解通路相关的基因信息,这表明S3-2-LX具备降解芳香族化合物的潜力。该研究结果与对Falsochrobactrum TDYN1[18]和Bacillus marsiflavi[27]菌株的研究结果相似,均从基因组水平和降解实验水平揭示了这些菌属的PAHs降解能力。PAHs的代谢途径主要包括羟基化、脱氢、苯环裂解和异构等步骤,将芳香族化合物分解为小分子脂肪酸,最终转化为H2O和CO2[20]。如,在S3-2-LX菌株中发现的2-氧葡萄糖酸盐/2-氧酸铁氧化还蛋白氧化还原酶亚基(korA /korB)、铁氧还蛋白亚硝酸盐还原酶基因(nirA)参与苯环上的双加氧反应,是PAHs降解的关键和限速步骤[28-29]。而加氧过后,在adhP、adhE、frmA等酶基因的催化下进行脱氢过程[30]。hmgA、catE、4-羟苯(基)丙酮酸双(加)氧酶基因(hppD)和4-羟基苯甲酸盐-3-单加氧酶基因(hpaB)等酶基因则催化苯环的裂解[29]。而praC、acd(脂酰辅酶A脱氢酶)、fadD(长链酰基辅酶A合酶)等酶基因可以催化开环后脂肪酸的降解过程[31]。此外,S3-2-LX菌株基因组的KEGG基因注释中还发现了ssuD、cypD_E等酶基因。ssuD或cypD_E能将烷烃末端的甲基催化氧化成羟基,转化为醇类,是烷烃降解的关键步骤和限速步骤[32]。adhP和adhE进一步将醇类氧化为相应的醛和脂肪酸[33]。此外,acd和fadD等酶基因可将脂肪酸β-氧化生成乙酰辅酶A,并通过进一步的三羧酸循环进行代谢,最终生成H2O和CO2[34]。这些基因的检出表明,S3-2-LX菌株也具备降解烷烃的潜力。
3.4 其他功能基因对S3-2-LX菌株的柴油降解能力的影响
通过基因组COG分析发现,S3-2-LX菌株包含转录调控因子LysR型、GntR型、EamA-like型、IcIr型、MarR型和AraC型家族调节因子。研究表明,LysR型转录调节因子是原核生物中广泛存在的转录调控因子之一,在调节芳香族化合物分解代谢、细胞运动和群体感应中的基因表达中起着关键作用[35]。而GntR型、EamA-like型、IcIr型、MarR型和AraC型家族调节因子也能够调节芳香族化合物的代谢过程[35-36]。这些基因的检出表明该菌株能快速地响应PAHs的胁迫,并对其进行降解代谢。此外,还发现了趋化性和细胞运动基因,包括编码鞭毛蛋白(如FliS、FlgC、FliE等)和趋化复合体蛋白(CheC、CheR、CheY、CheZ)的基因。有研究证明,趋化性和细胞运动基因能够有效提高柴油降解微生物对碳氢化合物的生物可利用效率[37-38],有助于提高微生物对柴油的降解效率。
4 结 论
本研究从土壤中分离得到柴油降解菌Bacillus S3-2-LX,发现其具有较强的PAHs降解能力。基因组测序和分析发现其具有柴油降解代谢基因、转录调控基因、趋化性和细胞运动基因、可移动遗传元件基因等,这些基因为该菌株的柴油降解能力提供了遗传基础。该菌株的分离和代谢途径的分析为柴油污染的生物修复提供了优质菌株来源和理论支撑。
参考文献:
[1] AHMED F,FAKHRUDDIN A.A review on environmental contamination of petroleum hydrocarbons and its biodegradation[J].International Journal of Environmental Sciences & Natural Resources,2018,11(3):63-69.
[2] GUO W,WANG X,LIU S,et al.Long-term petroleum hydrocarbons pollution after a coastal oil spill[J].Journal of Marine Science and Engineering,2022,10(10):1380.
[3] CZARNY J,STANINSKA P J,PIOTROWSKA C A,et al.Acinetobacter sp.as the key player in diesel oil degrading community exposed to PAHs and heavy metals[J].Journal of Hazardous Materials,2020,383:121168.
[4] ZHANG C,WU D,REN H.Bioremediation of oil contaminated soil using agricultural wastes via microbial consortium[J].Scientific Reports,2020,10(1):9188.
[5] 张胜,陈立,李政红,等.中原石油污染土壤原位微生物生态修复技术的应用[J].微生物学通报,2011,38(4):615-620.
[6] ACOSTA-GONZLEZ A,MARQUS S.Bacterial diversity in oil-polluted marine coastal sediments[J].Current Opinion in Biotechnology,2016,38:24-32.
[7] LEITE G G F,FIGUEIRA J V,ALMEIDA T C M,et al.Production of rhamnolipids and diesel oil degradation by bacteria isolated from soil contaminated by petroleum[J].Biotechnology Progress,2016,32(2):262-270.
[8] GAO J,MING J,XU M,et al.Isolation and characterization of a high-efficiency marine diesel oil-degrading bacterium[J].Petroleum Science,2021,18(2):641-653.
[9] KHAN A L,NUMAN M,BILAL S,et al.Mangrove’s rhizospheric engineering with bacterial inoculation improve degradation of diesel contamination[J].Journal of Hazardous Materials,2022,423:127046.
[10]OYEWOLE O A,RAJI R O,OKEKE S K,et al.Potential of fungi isolated from diesel contaminated soil to degrade diesel[J].Tanzania Journal of Science,2021,47(1):47-56.
[11]GARRIDO-SANZ D,REDONDO-NIETO M,GUIRADO M,et al.Metagenomic insights into the bacterial functions of a diesel-degrading consortium for the Rhizoremediation of diesel-polluted soil[J].Genes,2019,10(6):456.
[12]GRAN-SCHEUCH A,FUENTES E,BRAVO D M,et al.Isolation and characterization of phenanthrene degrading bacteria from diesel fuel-contaminated antarctic soils[J].Frontiers in Microbiology,2017,8:1634.
[13]SAKSHI,HARITASH A K.A comprehensive review of metabolic and genomic aspects of PAH-degradation[J].Archives of Microbiology,2020,202(8):2033-2058.
[14]ROJO F.Degradation of alkanes by bacteria[J].Environmental Microbiology,2009,11(10):2477-2490.
[15]吴慧君,宋权威,郑瑾,等.微生物降解石油烃的功能基因研究进展[J].微生物学通报,2020,47(10):3355-3368.
[16]HAMME J D V,SINGH A,WARD O P.Recent advances in petroleum microbiology[J]. Microbiology and Molecular Biology Reviews,2003,67(4):503-549.
[17]ZHANG X,XU D,ZHU C,et al.Isolation and identification of biosurfactant producing and crude oil degrading Pseudomonas aeruginosa strains[J].Chemical Engineering Journal,2012,209:138-146.
[18]陈冠益,陈红云,栗高源,等.一株石油降解菌 TDYN1 基因组测序及分析[J].生物学杂志,2023,40(1):34-39.
[19]徐靖,牛邦彦,张亚南,等.芽孢杆菌属Bacillus分类学研究进展[J].中国土壤与肥料,2022(12):225-237.
[20]CHOI E J,JIN H M,LEE S H,et al.Comparative genomic analysis and benzene,toluene,ethylbenzene,and o-,m-,and p-xylene (BTEX) degradation pathways of Pseudoxanthomonas spadix BD-a59[J].Applied and Environmental Microbiology,2013,79(2):663-671.
[21]曹军伟.深海多环芳烃降解菌Celeribacter indicus P73T降解荧蒽和菲的机制研究[D].哈尔滨:哈尔滨工业大学,2016.
[22]PARELLADA E A,IGARZA M,ISACC P,et al.Squamocin,an annonaceous acetogenin,enhances naphthalene degradation mediated by Bacillus atrophaeus CN4[J].Revista Argentina de Microbiologia,2017,49(3):282-288.
[23]LU Q,CHEN K,LONG Y,et al.Benzo(a)pyrene degradation by cytochrome P450 hydroxylase and the functional metabolism network of Bacillus thuringiensis[J].Journal of Hazardous Materials,2019,366:329-337.
[24]RABODONIRINA S,RASOLOMAMPIANINA R,KRIER F,et al.Degradation of fluorene and phenanthrene in PAHs-contaminated soil using Pseudomonas and Bacillus strains isolated from oil spill sites[J].Journal of Environmental Management,2019,232:1-7.
[25]MANDREE P,MASIKA W,NAICKER J,et al.Bioremediation of polycyclic aromatic hydrocarbons from industry contaminated soil using indigenous Bacillus spp.[J].Processes,2021,9(9):1606.
[26]GIUNTA M,BOSCO D L,LEONARDI G,et al.Estimation of gas and dust emissions in construction sites of a motorway project[J].Sustainability,2019,11(24):7218.
[27]SAEED M,ILYAS N,BIBI F,et al.Biodegradation of PAHs by Bacillus marsiflavi,genome analysis and its plant growth promoting potential[J].Environmental Pollution,2022,292:118343.
[28]KIM S,KWEON O,JONES R C,et al.Genomic analysis of polycyclic aromatic hydrocarbon degradation in Mycobacterium vanbaalenii PYR-1[J].Biodegradation,2008,19(6):859-881.
[29]LI X,QU C,BIAN Y,et al.New insights into the responses of soil microorganisms to polycyclic aromatic hydrocarbon stress by combining enzyme activity and sequencing analysis with metabolomics[J].Environmental Pollution,2019,255:113312.
[30]SIERRA-GARCA I N,ALVAREZ J C,DE VASCONCELLOS S P,et al.New hydrocarbon degradation pathways in the microbial metagenome from Brazilian petroleum reservoirs[J].PLos ONE,2014,9(2):e90087.
[31]ISMAIL W,GESCHER J.Epoxy coenzyme a thioester pathways for degradation of aromatic compounds[J].Applied and Environmental Microbiology,2012,78(15):5043-5051.
[32]MISHRA S,SINGH S N.Microbial degradation of n-hexadecane in mineral salt medium as mediated by degradative enzymes[J].Bioresource Technology,2012,111:148-154.
[33]PAL S,KUNDU A,BANERJEE T D,et al.Genome analysis of crude oil degrading Franconibacter pulveris strain DJ34 revealed its genetic basis for hydrocarbon degradation and survival in oil contaminated environment[J].Genomics,2017,109(5-6):374-382.
[34]HONG Y H,DENG M C,XU X M,et al.Characterization of the transcriptome of Achromobacter sp.HZ01 with the outstanding hydrocarbon-degrading ability[J].Gene,2016,584(2):185-194.
[35]DAS D,BARUAH R,ROY A S,et al.Complete genome sequence analysis of Pseudomonas aeruginosa N002 reveals its genetic adaptation for crude oil degradation[J].Genomics,2015,105(3):182-190.
[36]CAO J,LAI Q,YUAN J,et al.Genomic and metabolic analysis of fluoranthene degradation pathway in Celeribacter indicus P73T[J].Scientific Reports,2015,5:7741.
[37]范萌,黄升泉,李昱龙,等.芽孢杆菌鞭毛及其运动相关特性的研究进展[J].微生物学通报,2022,49(5):1832-1845.
[38]IBRAR M,KHAN S,HASAN F,et al.Biosurfactants and chemotaxis interplay in microbial consortium-based hydrocarbons degradation[J].Environmental Science and Pollution Research,2022,29(17):24391-24410.
Analysis on the Screening,Identification and Genome Sequencingof Diesel-degrading Strain Bacillus S3-2-LX
YE Chun-honga,YANG Rui-yub,PENG Chaoa,LU Lub
(a.College of Life Science,b.College of Environmental Science and Engineering,China West Normal University,Nanchong Sichuan 637009,China)
Abstract:Diesel pollution has posed serious threats to environmental biodiversity and human health.Bioremediation is an important approach to deal with diesel pollution.Taking diesel as the only carbon source,this study has isolated a strain from the soil of a garbage dump site in a university of Sichuan Province.The strain named S3-2-LX belongs to the Bacillus genus.Its thallus is rod-shaped,and its bacterial colony is oyster white and round.To verify the degradation capability of the strain towards polycyclic aromatic hydrocarbons (PAHs),a PAH degradation cultivation experiment is conducted and de novo sequencing is performed on the Illumina HiSeq 2000 platform.The results are as follows:strain S3-2-LX has exhibited high degradation rates of naphthalene (99.1%),phenanthrene (42.1%),and pyrene (29.2%) after 7 days of cultivation;the total sequencing length of S3-2-LX genome is 5 869 851 bp and a total of 53 Scaffolds are obtained,with a GC content of 34.89%;gene annotation by the COG and KEGG databases has identified 4 211 and 3 060 genes,respectively;S3-2-LX possesses pathways related to hydrocarbon metabolism and the ability to degrade alkane,dioxins,xylene,benzoic acid,and polycyclic aromatic hydrocarbons.The analysis on the physiological characteristics and metabolic pathways of S3-2-LX in this study has further revealed the key steps and mechanisms of its diesel degradation.
Keywords:diesel-degrading bacteria;isolation and identification;Bacillus;genome sequencing;gene annotation
