基于PI3K/Akt/mTOR信号通路的miR-24调控ox-LDL诱导的HUVECs自噬机制研究
2024-11-07杨鹏杨增艳翟阳周炜潜罗雪兰欧和生



【摘要】 目的 探讨miR-24对氧化低密度脂蛋白(ox-LDL)诱导下的人脐静脉内皮细胞(HUVECs)自噬的影响及其相关机制,为进一步阐明miR-24在动脉粥样硬化(AS)中的作用提供理论依据。
方法 采用实时荧光定量PCR(qRT-PCR)检测miR-24的表达;应用蛋白免疫印迹法(western blot)和qRT-PCR法检测Beclin-1、LC3Ⅰ/LC3Ⅱ、p-mTOR、p-PI3K、p-Akt的蛋白和mRNA表达水平;应用透射电子显微镜技术检测细胞的自噬小体生成情况;应用四甲基偶氮唑盐(MTT)法、细胞划痕法、Caspase-3比色法和Hoechst 33258染色法分别检测细胞活性、迁移和凋亡情况。
结果 应用ox-LDL诱导HUVECs后,发现HUVECs中miR-24的表达显著降低(P<0.05)。miR-24过表达可明显抑制ox-LDL诱导的HUVECs自噬(P<0.05),而miR-24低表达则会增加ox-LDL诱导的HUVECs自噬(P<0.05)。miR-24过表达可显著降低Beclin-1的表达水平,上调LC3Ⅰ/LC3Ⅱ的水平(P<0.05),同时,miR-24过表达可显著促进p-PI3K、p-Akt和p-mTOR的表达(P<0.05)。此外,miR-24过表达显著抑制HUVECs的活力和迁移,增加Caspase-3活性并促进其凋亡(P<0.05)。
结论 miR-24的过表达可激活PI3K/Akt/mTOR信号通路而降低ox-LDL诱导的HUVECs的自噬水平并促进其凋亡,miR-24可能成为AS的潜在治疗新靶点。
【关键词】 氧化低密度脂蛋白;人脐静脉内皮细胞;PI3K/Akt/mTOR信号通路;miR-24;自噬;凋亡
中图分类号:R36.1+2;R544 文献标志码:A DOI:10.3969/j.issn.1003-1383.2024.09.002
Study on the regulation of ox-LDL induced autophagy mechanism in HUVECs by miR-24 based on PI3K/Akt/mTOR signaling pathway
YANG Peng, YANG Zengyan, ZHAI Yang, ZHOU Weiqian, LUO Xuelan, OU Hesheng▲
(Department of Science and Technology, Guangxi International Zhuang Medical Hospital, Nanning 530201, Guangxi, China)
【Abstract】 Objective To investigate the effect of miR-24 on autophagy in human umbilical vein endothelial cells (HUVECs) induced by oxidized low-density lipoprotein (ox-LDL) and its related mechanism, so as to provide a theoretical basis for further elucidating the role of miR-24 in atherosclerosis (AS).
Methods Real-time fluorescence quantitative PCR (qRT-PCR) was used to detect the expression of miR-24; western blot and qRT-PCR were applied to detect the protein and mRNA expression levels of Beclin-1, LC3Ⅰ/LC3Ⅱ, p-mTOR, p-PI3K, and p-Akt; transmission electron microscopy was used to measure the formation of autophagosomes in cells; MTT assay, cell scratch assay, Caspase-3 colorimetric assay, and Hoechst 33258 staining were used to assess cell viability, migration, and apoptosis, respectively.
Results After inducing HUVECs with ox-LDL, it was found that the expression of miR-24 in HUVECs was significantly reduced (P<0.05). Overexpression of miR-24 significantly inhibited autophagy induced by ox-LDL in HUVECs (P<0.05), while downregulation of miR-24 increased autophagy induced by ox-LDL in it (P<0.05). The overexpression of miR-24 could significantly reduce the expression levels of Beclin-1 and upregulate the levels of LC3Ⅰ/LC3Ⅱ(P<0.05). At the same time, the overexpression of miR-24 could significantly promote the expressions of p-PI3K, p-Akt, and p-mTOR (P<0.05). Furthermore, overexpression of miR-24 significantly inhibited the viability and migration of HUVECs, increased the activity of Caspase-3, and promoted apoptosis (P<0.05).
Conclusion Overexpression of miR-24 can activate PI3K/Akt/mTOR signaling pathway, reduce the level of autophagy induced by ox-LDL in HUVECs, and promote apoptosis, which miR-24 may become a potential new therapeutic target for AS.
【Keywords】 oxidized low-density lipoprotein (ox-LDL); human umbilical vein endothelial cells (HUVECs); PI3K/Akt/mTOR signaling pathway; miR-24; autophagy; apoptosis
动脉粥样硬化(atherosclerosis, AS)的病理过程十分复杂,血管内皮细胞(vascular endothelial cells, VECs)损伤、自噬功能障碍和miRNA的异常表达等多种因素都参与了AS的发生发展过程[1-2]。氧化低密度脂蛋白(oxidized low-density lipoprotein, ox-LDL)是一种促AS发展的关键因子[3],其可导致VECs损伤、内皮细胞自噬功能障碍和凋亡,这有助于促进AS的发生发展[4]。
自噬是一种维护细胞内环境和组织稳态的机制。在某些应激条件下,细胞需要降解胞内不必要的蛋白质和细胞器来维持自身生存及稳态,这一过程可通过自噬来实现。当VECs损伤后,可诱发自噬以保护细胞,而当自噬诱发失败或被抑制时,内皮的完整性被破坏而促进局部脂质沉积,导致AS、斑块不稳定、急性血管闭塞甚至猝死[5]。自噬调节机制复杂,目前被认为与多条信号通路有关,其中PI3K/Akt/mTOR信号通路是典型的自噬信号传导途径,该通路可能参与AS自噬的诱导[6-7],但其具体的分子机制还需进一步探索。AS的发生还受到基因表达的调控,在基因的转录过程中,microRNAs(miRNAs)的差异性表达与AS的发生发展密切相关。有研究表明,部分miRNAs可通过影响VECs的增殖、迁移、自噬和凋亡来促进AS的进展[8],如位于人类9号染色体上的miR-24,其成熟序列在心血管疾病中起着至关重要的作用[9]。在前期的研究中,已发现miR-24可显著抑制人脐静脉内皮细胞(humanumbilical vein endothelial cell, HUVECs)自噬及增殖[10]。因此,本研究为进一步探讨miR-24与HUVECs自噬之间的相关分子机制及其是否对AS有潜在作用,将采用ox-LDL诱导HUVECs自噬并体外模拟AS模型,以PI3K/Akt/mTOR为切入点探究miR-24调控HUVECs自噬的相关分子机制,进一步探讨miR-24通过调控自噬在AS中的潜在价值,为AS提供潜在的治疗靶点。
1 材料与方法
1.1 细胞培养与ox-LDL诱导HUVECs
HUVECs购自中国科学院细胞库,用含15%新生胎牛血清(15% FBS)的DMEM培养液,加入青霉素100 IU/mL和链霉素0.1 mg/mL,置于37 ℃、5%CO2饱和湿度培养箱中培养。将实验HUVECs分别用0、25、50、100和200 g/mL浓度的ox-LDL处理0、6、12、24和48小时,检测miR-24在ox-LDL诱导的HUVECs中表达情况。
1.2 细胞转染
委托上海吉凯基因化学技术有限公司,设计合成并得到miR-24高表达、anti-miR-24和miR-NC三种载体质粒,见表1。取对数生长期细胞,分别以每孔5×104个细胞接种到6孔板,正常静置培养24 h,按照转染试剂说明书要求,将以上3种质粒分别转染至HUVECs中,继续置于培养箱中孵育48 h,在倒置荧光显微镜下观察荧光表达情况,加入10 μg/mL嘌呤霉素进行6sQQ26LA1K0cH66ynfgkDTh+0xWh2F+Sx7YY7msOMdI=筛选。
1.3 qRT-PCR检测miR-24及其他基因mRNA的表达
按TRLzol试剂说明提取HUVECs总RNA,然后使用miRNA反转录试剂盒合成互补DNA,使用SYBR Green PCR Master Mix试剂(Qiagen)进行qPCR分析。miRNA和mRNA的表达以U6或GAPDH为内参,并采用2-△△Ct进行数据分析处理。本实验中使用的引物序列见表2。
1.4 MTT法检测HUVECs的活性
将HUVECs以5×103个/孔的密度接种到96孔板中,置于培养箱中培养,待细胞贴壁后向每个孔中加入MTT试剂10 L,继续孵育4小时,然后除去上清液,再加入150 L DMSO,振荡10 s,最后用酶联免疫检测仪(Model-450)测定490 nm波长下的吸光度(A490nm),通过测试孔的吸光度减去空白孔的光密度表示细胞的存活/增殖。
1.5 划痕试验检测HUVECs的迁移
将HUVECs以5×104个/孔的密度接种到24孔板上并孵育至融合(每组3次重复),随后用枪头垂直于培养板底部划“一”字痕,PBS洗去被划下的细胞,再加入无血清培养基,在划痕后0和24小时的时间点对HUVECs进行拍照。计算公式为:细胞迁移率(%)=(A0-An)/A0×100,其中A0代表初始划痕面积,An代表时间计量点处划痕面积。
1.6 Caspase-3活性测定检测HUVECs的凋亡
按照说明书上的步骤,将细胞裂解液与酶特异性底物(100 uM)在37 ℃下孵育4小时,然后使用酶标仪在405 nm处测量吸光度。
1.7 Hoechst 33258染色法检测HUVECs的凋亡
将细胞以5×104个/孔的密度接种到12孔板中。培养24小时待细胞贴壁后,用冷PBS冲洗细胞2次,然后用4%多聚甲醛溶液将贴壁细胞在4 ℃中固定10 min,随后再次用冷PBS冲洗2遍,接着加入Hoechst 33258试剂(0.2 mM),并在室温避光下培养10 min,应用荧光显微镜观察细胞核形态变化。细胞核固缩或碎裂成致密颗粒状,呈亮蓝色荧光的细胞为凋亡细胞。
1.8 透射电子显微镜法观察各组HUVECs的自噬小体
将细胞漂洗后置于2.5%戊二醛溶液中,避光并置于4 ℃冰箱中固定过夜。次日用PBS漂洗3次后,锇酸溶液固定2 h。重复用PBS溶液漂洗3次,丙酮梯度脱水,纯树脂渗透过夜,环氧树脂包埋,烘箱聚合。超薄切片,醋酸铀、柠檬酸铅双染色,使用透射电子显微镜(H-7650)观察并拍照。
1.9 western blot法检测Beclin-1、LC3Ⅰ/LC3Ⅱ、p-mTOR、mTOR、p-PI3K、PI3K、p-Akt和Akt的蛋白表达
将实验细胞裂解取上清液,用BCA标准曲线蛋白定量,分别取各组30 μg蛋白进行SDS-PAGE电泳,经电泳后转移至PVDF膜上。封闭后加入一抗孵育置于4 ℃冰箱过夜,次日用TBST冲洗3次,然后加入荧光二抗孵育1 h后反复冲洗3次。用双色荧光扫描成像系统分析蛋白电泳条带的灰度值,以β-actin为内参,蛋白相对表达量=目的蛋白灰度值/β-actin灰度值。实验重复3次,取平均值。
1.10 统计学方法
采用SPSS 25.0软件进行统计分析,所有数据用(±s)表示。多组间比较用单因素方差分析,组内两两比较用SNK-q检验,检验水准:α=0.05,双侧检验,实验独立重复三次。
2 结 果
2.1 miR-24在ox-LDL诱导的HUVECs中表达降低
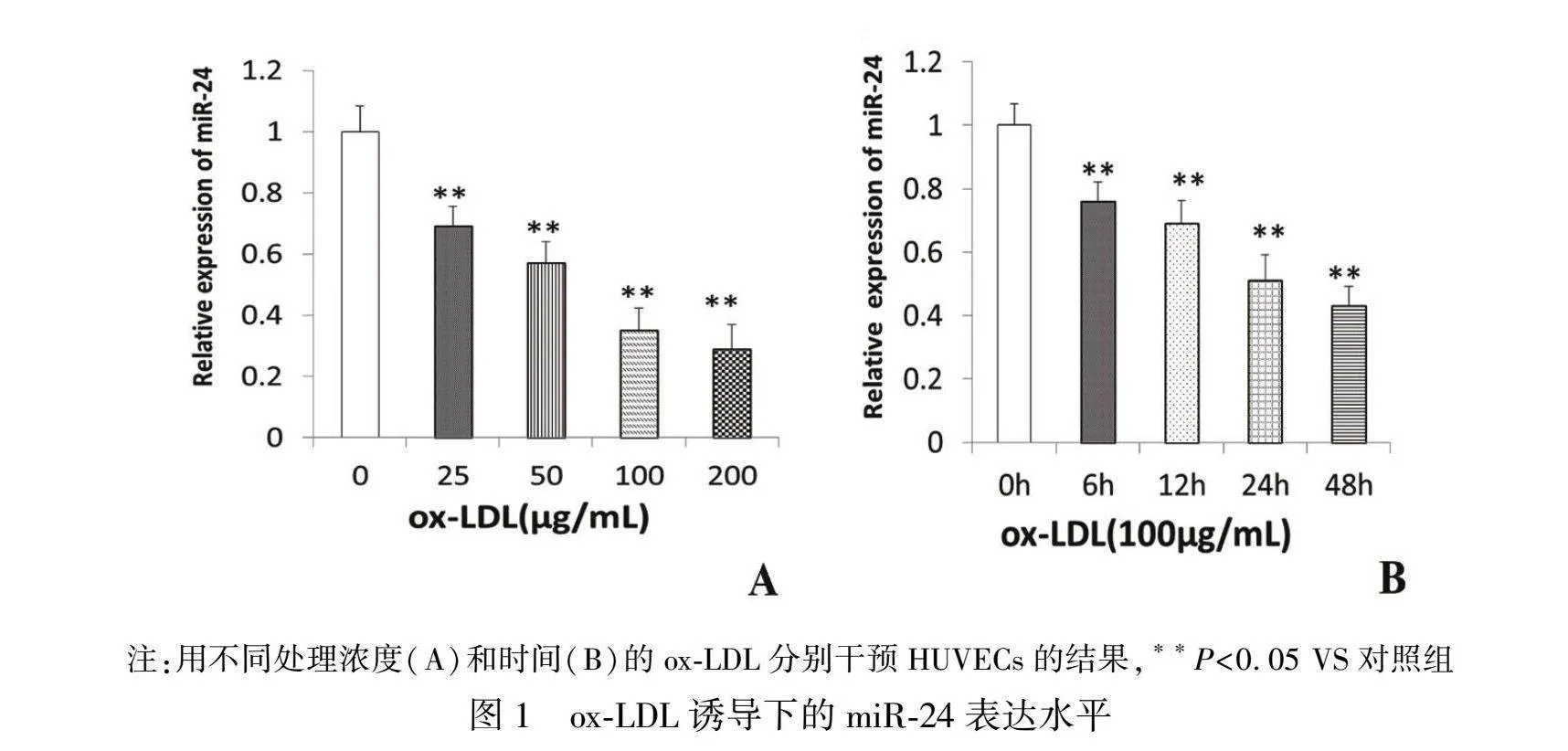
为探讨miR-24在ox-LDL诱导的HUVECs中表达的水平,本研究采用qRT-PCR检测miR-24的表达。如图1A和1B所示,miR-24在ox-LDL诱导的HUVECs中的表达呈时间和剂量依赖性下调,用100 g/mL的ox-LDL干预HUVECs 24 h后,miR-24表达水平下调约50%(P<0.05),提示ox-LDL可明显降低HUVECs中miR-24表达水平,miR-24的降低可能与ox-LDL诱导HUVECs损伤有关。
2.2 ox-LDL可诱导HUVECs自噬
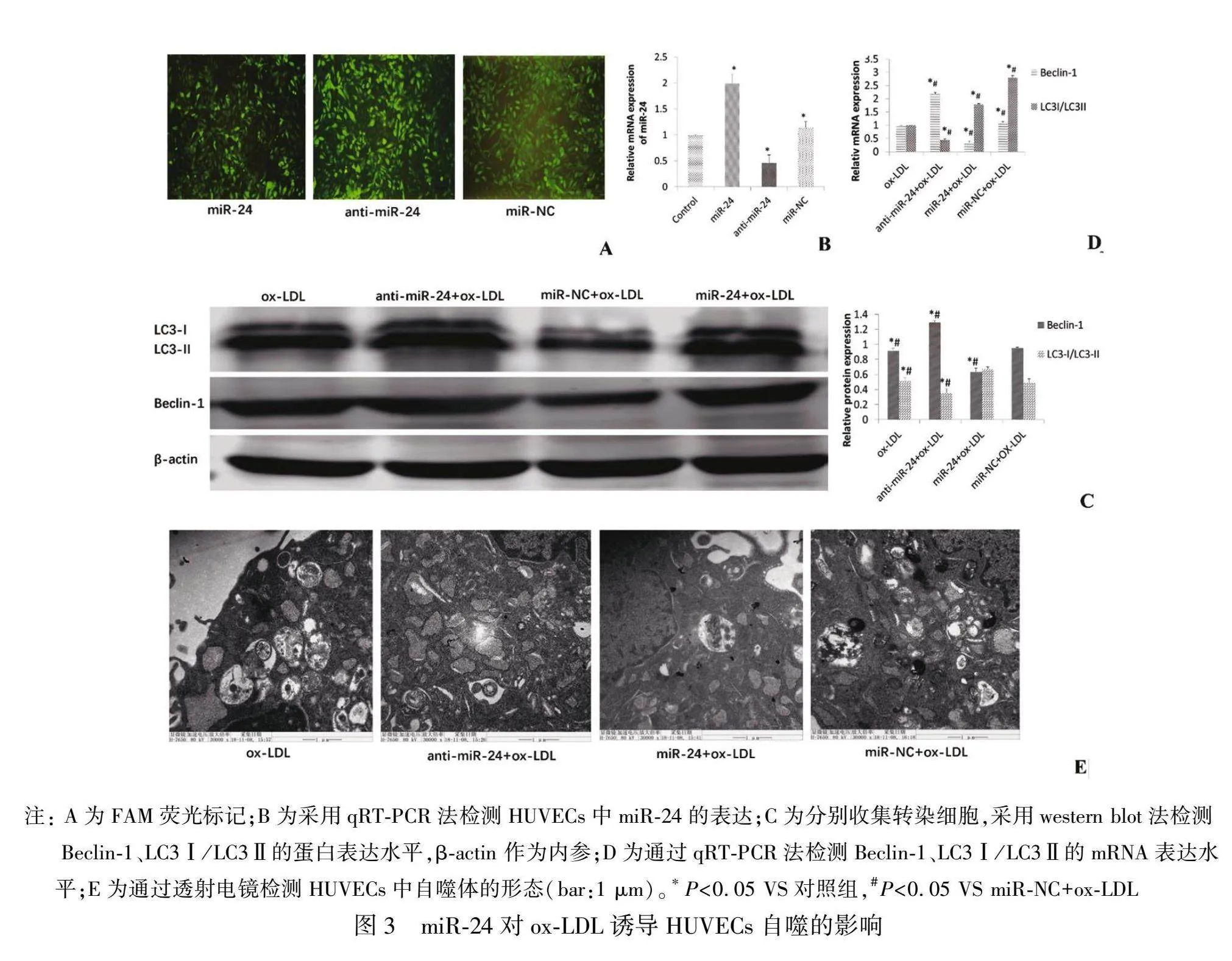
为研究ox-LDL诱导HUVECs自噬的情况,本研究用100 g/mL的ox-LDL干预 HUVECs 6 h建立自噬模型。通过透射电镜法观察建模前后自噬小体生成情况,结果显示与正常对照组比较,ox-LDL组中HUVECs的细胞质中含有更多具有双膜结构的自噬小体和含细胞器的空泡型自噬溶酶体(图2A)。此外,通过western blot和qRT-PCR法检测发现,ox-LDL能显著上调自噬标志基因Beclin-1的表达水平,并下调LC3Ⅰ/LC3Ⅱ的水平(P<0.05)。见图2B和2C。以上结果表明,用100 g/mL的ox-LDL干预 HUVECs 6 h可诱导HUVECs自噬。
2.3 miR-24的过表达可抑制ox-LDL诱导的HUVECs自噬
为了研究miR-24对ox-LDL诱导自噬的调控作用,本研究构建高表达miR-24质粒、anti-miR-24质粒和miR-NC质粒,并分别转染至经ox-LDL诱导过的HUVECs中,采用qRT-PCR方法检测转染效率(图3B),与阴性对照组(miR-NC)和ox-LDL组比较vvjB2cPuCvjd5Aes4kQAHg==,miR-24组中的miR-24表达水平显著升高(P<0.05),anti-miR-24组中的miR-24表达水平显著降低(P<0.05),阴性对照组与ox-LDL组中的miR-24表达水平无明显变化,差异无统计学意义(P>0.05)。此外,FAM荧光标记表明(图3A),HUVECs中的质粒转染率达90%以上。这些结果表明,高表达miR-24质粒、anti-miR-24质粒和miR-NC质粒均成功转染至ox-LDL诱导的HUVECs中。通过western blot和qRT-PCR法检测各组Beclin-1、LC3Ⅰ/LC3Ⅱ的蛋白和mRNA表达水平,结果显示(图3C和3D):与miR-NC组比较,miR-24高表达组的Beclin-1蛋白和mRNA表达水平显著降低,而LC3Ⅰ/LC3Ⅱ的蛋白和mRNA表达水平明显升高(P<0.05),anti-miR-24组的Beclin-1蛋白和mRNA表达水平显著升高,而LC3Ⅰ/LC3Ⅱ的蛋白和mRNA表达水平明显降低(P<0.05),提示miR-24可抑制ox-LDL诱导的HUVECs自噬。为了进一步证明miR-24对ox-LDL诱导的HUVECs自噬的调控作用,我们通过透射电镜观察各组自噬小体的生成情况(图3E),与miR-NC组相比,miR-24高表达组细胞质中可见数个散在于细胞质中的双层膜结构的自噬小体及内含细胞器的空泡状自噬溶酶体,而anti-miR-24组细胞质中可见较多的自噬小体,进一步证明miR-24可抑制ox-LDL诱导的HUVECs自噬。
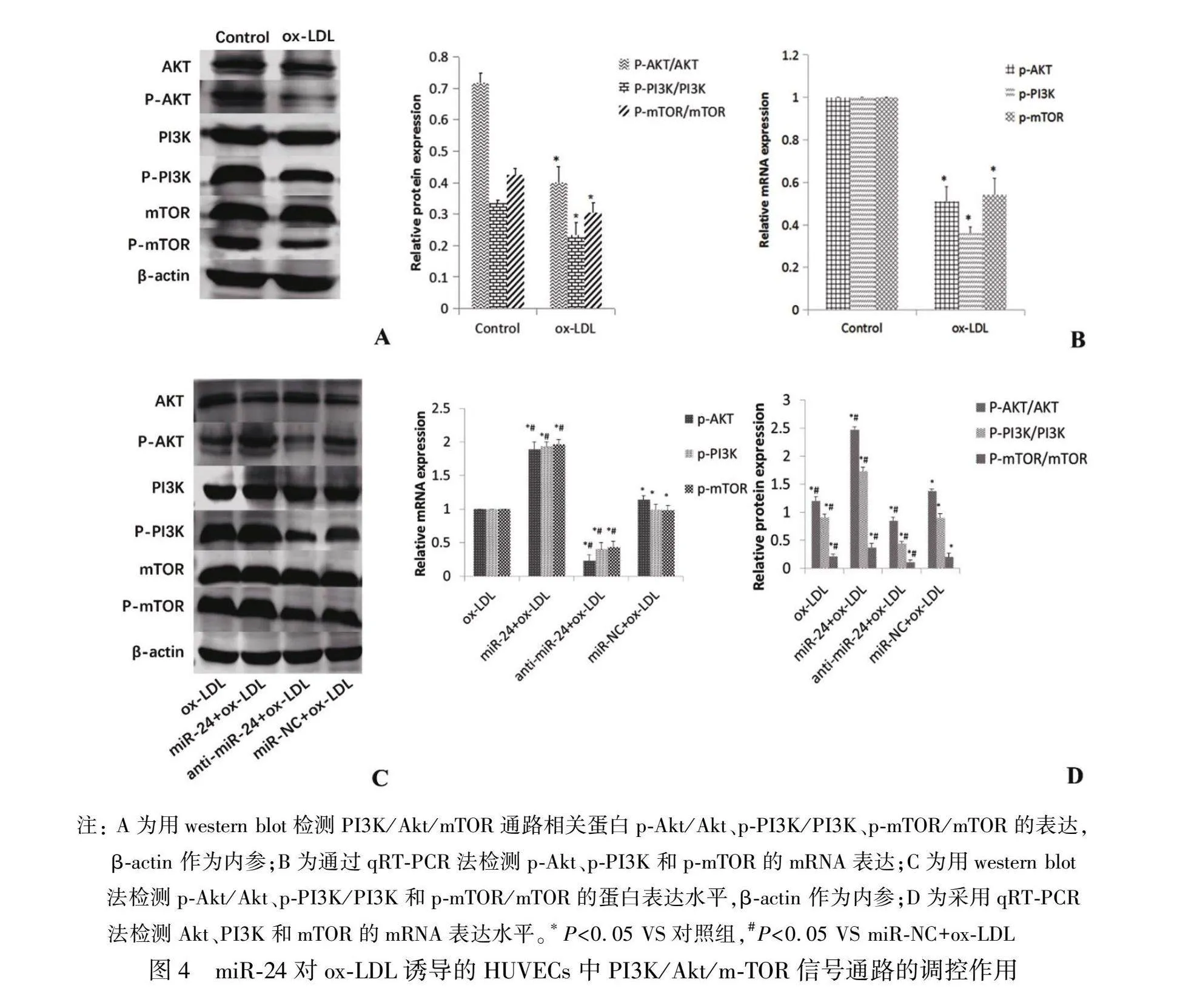
2.4 miR-24通过激活PI3K/Akt/mTOR通路抑制自噬
为了进一步探讨miR-24抑制ox-LDL诱导HUVECs自噬的潜在机制,本研究采用western blot和qRT-PCR检测与PI3K/Akt/mTOR通路相关的蛋白和mRNA的表达。结果发现:与正常对照组相比,ox-LDL明显降低了磷酸化PI3K(p-PI3K)、磷酸化Akt(p-Akt)、磷酸化mTOR(p-mTOR)蛋白和mRNA的表达(P<0.05),见图4A和4B。与miR-NC组相比,miR-24高表达组中p-PI3K、p-Akt、p-mTOR蛋白和mRNA的表达显著上调(P<0.05),而anti-miR-24组中p-PI3K、p-Akt、p-mTOR蛋白和mRNA的表达显著下调(P<0.05),见图4C和4D,提示过表达miR-24可通过激活PI3K/Akt/mTOR通路抑制ox-LDL诱导的HUVECs自噬。
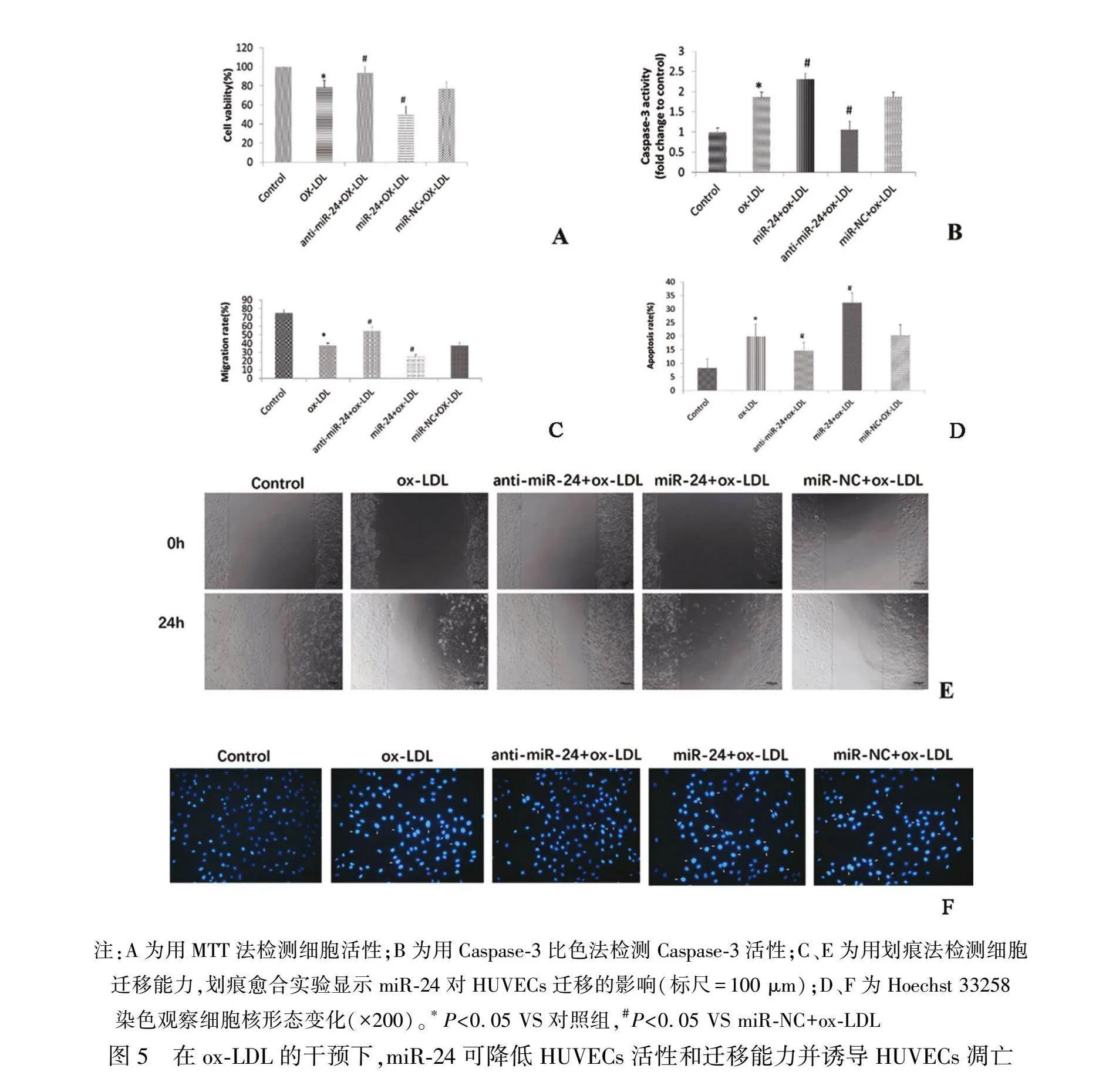
2.5 miR-24可降低HUVECs活性和迁移能力并诱导HUVECs凋亡
本研究采用MTT检测HUVECs增殖情况(图5A),与正常对照组比较,ox-LDL组HUVECs增殖明显受到抑制(P<0.05),阴性对照组与ox-LDL组比较无明显变化(P>0.05)。与阴性对照组相比,miR-24高表达组的HUVECs增殖受到抑制(P<0.05),anti-miR-24组的HUVECs增殖增加(P<0.05)。本研究采用划痕试验进一步检测miR-24对HUVECs迁移能力的影响(图5C和5E),与正常对照组相比,ox-LDL组HUVECs迁移能力明显受到抑制(P<0.05),阴性对照组与ox-LDL组比较无明显变化(P>0.05)。与阴性对照组相比,miR-24高表达组HUVECs的迁移能力受到抑制(P<0.05),而anti-miR-24组细胞的迁移能力得到了提高(P<0.05)。
为证实miR-24是否参与调控HUVECs的凋亡,本研究采用Caspase-3比色法和Hoechst 33258染色法检测分析HUVECs凋亡的变化。Caspase-3比色法结果显示(图5B):与正常对照组相比,ox-LDL组的Caspase-3活性明显增加(P<0.05);与阴性对照组相比,miR-24高表达组的Caspase-3的活性更强(P<0.05),而anti-miR-24组的Caspase-3活性明显降低(P<0.05)。Hoechst 33258双染色结果显示(图5D和5F):正常对照组HUVECs的细胞核形态正常,结构清晰,呈弥散均匀的浅蓝色荧光,未发生凋亡,而与正常对照组相比,ox-LDL组的HUVECs的细胞核固缩或碎裂成致密颗粒状,呈亮蓝色荧光,凋亡现象明显(P<0.05);与阴性对照组相比,miR-24过表达能显著促进ox-LDL诱导的HUVECs凋亡(P<0.05),而anti-miR-24能显著减弱ox-LDL诱导的HUVECs凋亡(P<0.05)。以上结果提示,miR-24的过表达能降低HUVECs的增殖和迁移能力并促进其凋亡,而下调miR-24水平则会增加HUVECs增殖和迁移能力并抑制其凋亡。
3 讨 论
本项目通过研究miR-24调控HUVECs自噬的潜在分子机制,进一步探讨miR-24通过调控自噬在AS中的潜在价值,主要有以下两个发现:(1)过表达miR-24可明显抑制ox-LDL诱导的HUVECs自噬并抑制HUVECs的增殖和迁移能力,进而发挥其在 AS 中的作用。(2)过表达miR-24可通过激活PI3K/Akt/mTOR信号通路从而降低HUVECs自噬水平并促进其凋亡。
AS是导致血管疾病死亡的主要原因之一[11]。ox-LDL是一种促AS发展的关键因子,可诱导VECs损伤进而导致AS的发生[12]。AS的重要特征之一为VECs的凋亡,其机制为通过ox-LDL降低Caspase-3活性而促进VECs的凋亡[13]。本研究中,XUJ4Epn+iav3/j7vsNzXkg==我们通过ox-LDL诱导HUVECs模拟体外AS模型,发现ox-LDL能显著降低HUVECs的活力,并增强Caspase-3的活性而促进HUVECs凋亡。还有其他研究表明,细胞的自噬在脂质代谢中发挥着重要的作用[14],其异常可导致细胞代谢紊乱而加速AS的发生[15]。在自噬过程中,一种以细胞质形式的LC3(LC3-Ⅰ)会被偶联形成LC3-磷脂酰乙醇胺偶联物(LC3-Ⅱ)募集到自噬体膜中,而此过程中LC3-Ⅱ始终稳定地保留在自噬体膜上直到与溶酶体融合,因此可作为自噬体的标记蛋白[16]。Beclin-1是磷脂酰肌醇-3-激酶复合物的组成部分,参与自噬体的形成[17],因此这两种蛋白质可以被看作是自噬活性的标志。ZHANG等[18]揭示了ox-LDL通过LC3/beclin-1途径激活HUVECs自噬。同样,我们的研究也发现,ox-LDL能上调自噬标志基因Beclin-1的表达水平,并下调LC3Ⅰ/LC3Ⅱ的表达水平,进而诱发HUVECs自噬(图2),这些结果表明ox-LDL通过LC3/Beclin1途径激活HUVECs的自噬。
此外,近年来越来越多的研究[19-21]表明,miRNAs参与细胞自噬和凋亡的调节,并在AS的发展中发挥重要作用,如通过抑制PI3K/Akt/mTOR通路恢复自噬通量来缓解ox-LDL诱导的HUVECs损伤[22]。miR-106b则通过靶向PTEN并可能激活PI3K/AKT信号通路,抑制了AS中内皮细胞的凋亡[23]。以上研究提示,miRNAs在HUVECs自噬和凋亡过程中扮演着重要角色,并与AS的发生发展关系密切。本课题组的前期研究发现,miR-24能够显著抑制HUVECs的增殖和迁移[10],在本研究中,我们首先在HUVECs中转染过表达miR-24的质粒,再用ox-LDL诱导HUVECs,通过MTT实验和Hoechst 33258染色检测发现,miR-24的过表达会增加ox-LDL诱导的HUVECs凋亡(图5A、5D、5F)。此外,我们还发现miR-24过表达可增加Caspase-3活性(图5B)并下调Beclin-1的蛋白表达,同时上调LC3Ⅰ/LC3Ⅱ的蛋白表达(图3C)。这些结果表明,miR-24能够加剧ox-LDL诱导的HUVEC的损伤,而这与其调节HUVECs的自噬和凋亡有关。而在自噬信号通路中,PI3K/Akt/mTOR信号通路最具有代表性,它在细胞存活、增殖、凋亡和自噬等多种细胞功能中扮演着重要的角色[24-26]。LI等[27]研究发现,通过抑制PI3K/AKT/mTOR信号通路调节巨噬细胞自噬,促进巨噬细胞向M2表型的极化,可减少动脉粥样硬化斑块损伤,改善血脂代谢和炎症水平。FENG等[28]报道可通过抑制PI3K来调节巨噬细胞中的AKT/mTOR和NF-κB信号通路的激活,减少炎症及与之相关的淋巴管生成,从而对动脉粥样硬化展现出全面的保护效果。这些研究表明PI3K/Akt/mTOR信号通路与细胞自噬和凋亡关系紧密。我们的研究结果显示,miR-24能够显著促进p-PI3K、p-Akt和p-mTOR的表达,这表明miR-24可通过激活PI3K/Akt/mTOR通路抑制ox-LDL诱导的HUVEC自噬并加剧细胞凋亡。
在本研究中,我们仅仅对miR-24在体外细胞实验中的调控作用及机制进行了初步探索,对于如何更好地揭示miR-24、自噬和细胞凋亡之间的关系仍需通过动物实验进行更深层次的验证。
综上所述,本研究结果表明了miR-24可通过激活PI3K/Akt/mTOR信号通路以抑制ox-LDL诱导的自噬,并促进HUVECs凋亡,这可能加重血管内皮细胞损伤,促进AS的发生发展。但是它们之间是否存在靶向调控关系还需要进一步的实验验证,随着进一步的体内验证和传递系统的优化,外源性anti-miR-24有望成为治疗AS的一种有前景的分子药物。
参 考 文 献
[1] GORABI A M, GHANBARI M, SATHYAPALAN T, et al. Implications of microRNAs in the pathogenesis of atherosclerosis and prospects for therapy[J]. Curr Drug Targets, 2021,22(15):1738-1749.
[2] NI D, MO Z C, YI G H. Recent insights into atherosclerotic plaque cell autophagy[J]. Exp Biol Med, 2021,246(24):2553-2558.
[3] PERROTTA I. The microscopic anatomy of endothelial cells in human atherosclerosis: focus on ER and mitochondria[J]. J Anat, 2020, 237(6):1015-1025.
[4] ZHENG J, LU C Z. Oxidized LDL causes endothelial apoptosis by inhibiting mitochondrial fusion and mitochondria autophagy[J]. Front Cell Dev Biol, 2020TDu2jSUH7iyHsVuMpO0i77D7YYHM4xpz9t5sUs/LrUs=,8:600950.
[5] ZHU Z S, LI J Y, ZHANG X R. Salidroside protects against ox-LDL-induced endothelial injury by enhancing autophagy mediated by SIRT1-FoxO1 pathway[J]. BMC Complementary Altern Med, 2019,19(1):111.
[6] FANG S H, WAN X, ZOU X Y, et al. Arsenic trioxide induces macrophage autophagy and atheroprotection by regulating ROS-dependent TFEB nuclear translocation and AKT/mTOR pathway[J]. Cell Death Dis, 2021,12(1):88.
[7] WAN Q, YANG M, LIU Z Y, et al. Atmospheric fine particulate matter exposure exacerbates atherosclerosis in apolipoprotein E knockout mice by inhibiting autophagy in macrophages via the PI3K/Akt/mTOR signaling pathway[J]. Ecotoxicol Environ Saf, 2021,208:111440.
[8] LI Z Y, ZHAO Y D, SUGURO S, et al. MicroRNAs regulate function in atherosclerosis and clinical implications[J]. Oxid Med Cell Longev, 2023,2023:2561509.
[9] YALIM Z, ONRAT S T, DURAL I E, et al. Could aneurysm and atherosclerosis-associated microRNAs (miR 24-1-5p,miR 34a-5p,miR 126-5p,miR 143-5p,miR 145-5p) also be associated with coronary artery ectasia?[J]. Genet Test Mol Biomarkers, 2023,27(9):290-298.
[10] ZHANG W Y, YAN L M, LI Y M, et al. Roles of miRNA-24 in regulating endothelial nitric oxide synthase expression and vascular endothelial cell proliferation[J]. Mol Cell Biochem, 2015,405(1/2):281-289.
[11] RUIZ-LEN A M, LAPUENTE M, ESTRUCH R, et al. Clinical advances in immunonutrition and atherosclerosis:a review[J]. Front Immunol, 2019,10:837.
[12] FANG H R, BO T Z, ZI X L, et al. Sophocarpine exert protective effect against ox-LDL-induced endothelial damage via regulating NF-κB signaling pathway[J]. Biosci Biotechnol Biochem, 2020,84(10):2104-2112.
[13] YANG S, ZHANG W, XUAN L L, et al. Akebia Saponin D inhibits the formation of atherosclerosis in ApoE-/- mice by attenuating oxidative stress-induced apoptosis in endothelial cells[J]. Atherosclerosis, 2019,285:23-30.
[14] CHAKRAVARTI B, AKHTAR SIDDIQUI J, ANTHONY SINHA R, et al. Targeting autophagy and lipid metabolism in cancer stem cells[J]. Biochem Pharmacol, 2023,212:115550.
[15] LIU S Z, YAO S J, YANG H, et al. Autophagy: regulator of cell death[J]. Cell Death Dis, 2023,14:648.
[16] PEA-MARTINEZ C, RICKMAN A D, HECKMANN B L. Beyond autophagy: LC3-associated phagocytosis and endocytosis[J]. Sci Adv, 2022,8(43):eabn1702.
[17] XU H D, QIN Z H. Beclin 1, bcl-2 and autophagy[J]. Adv Exp Med Biol, 2019,1206:109-126.
[18] ZHOU Y H, TANG Y Z, GUO L Y, et al. Overexpression of sFlt-1 represses ox-LDL-induced injury of HUVECs by activating autophagy via PI3K/AKT/mTOR pathway[J]. Microvasc Res, 2022,139:104252.
[19] ZHAO Y Y, WANG Z, ZHANG W H, et al. MicroRNAs play an essential role in autophagy regulation in various disease phenotypes[J]. Biofactors, 2019,45(6):844-856.
[20] LIU Y, SONG J W, LIN J Y, et al. Roles of microRNA-122 in cardiovascular fibrosis and related diseases[J]. Cardiovasc Toxicol, 2020,20(5):463-473.
[21] TABAEI S, TABAEE S S. Implications for microRNA involvement in the prognosis and treatment of atherosclerosis[J]. Mol Cell Biochem, 2021,476(3):1327-1336.
[22] THEOFILIS P, OIKONOMOU E, VOGIATZI G, et al. The role of microRNA-126 in atherosclerotic cardiovascular diseases[J]. Curr Med Chem, 2023,30(17):1902-1921.
[23] ZHANG Y Q, WANG L, XU J, et al. Up-regulated miR-106b inhibits ox-LDL-induced endothelial cell apoptosis in atherosclerosis[J]. Rev Bras De Pesquisas Med E Biol, 2020,53(3):e8960.
[24] ALZAHRANI A S. PI3K/Akt/mTOR inhibitors in cancer:at the bench and bedside[J]. Semin Cancer Biol, 2019,59:125-132.
[25] LIU X R, ZHANG L, YANG L C, et al. MiR-34a/c induce caprine endometrial epithelial cell apoptosis by regulating circ-8073/CEP55 via the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways[J]. J Cell Physiol, 2020,235(12):10051-10067.
[26] SU X, SHEN Z, YANG Q, et al. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms[J]. Theranostics, 2019,9(15):4461-4473.
[27] LI P, LI H, LI X, et al. San Jie Tong Mai Fang protects against atherosclerosis progression by regulating macroautophagy through the PI3K/AKT/mTOR signaling pathway[J]. J Cardiovasc Pharmacol, 2023,82(4):333-343.
[28] FENG X T, DU M, LI S J, et al. Hydroxysafflor yellow A regulates lymphangiogenesis and inflammation via the inhibition of PI3K on regulating AKT/mTOR and NF-κB pathway in macrophages to reduce atherosclerosis in ApoE-/- mice[J]. Phytomedicine, 2023,112:154684.
(收稿日期:2024-05-08 修回日期:2024-08-22)
