基于加权基因共表达网络和癌症基因组图谱临床数据分析并鉴定肝细胞癌的Hub基因研究
2024-08-06陈超陈天翔刘钱伟张秩王欢欢吴平平高磊于照祥


【摘要】 背景 肝细胞癌(HCC)是全球常见的癌症相关死亡的第三大原因,约占所有原发性肝癌病例的90%,其复发率和死亡率较高,目前发生的分子机制仍不清楚。目的 探索HCC潜在的分子机制,发掘新的生物标志物。方法 从TCGA数据库下载RNA-seq表达数据和临床相关信息,通过差异基因表达分析正常肝脏组织与HCC组织的差异基因;对差异表达基因进行富集分析;基于TCGA中HCC的基因表达数据概况,使用WGCNA R包建立共表达网络,进行加权基因共表达网络分析(WGCNA),选择具有临床意义的模块,并筛选候选Hub基因;进一步分析候选Hub基因在HCC组织和正常肝脏组织显著差异表达、与HCC患者总体生存期和无病生存期是否显著相关,最终确定Hub基因;通过人类蛋白质图谱数据库对Hub基因蛋白表达进行验证。结果 本研究的基因表达数据来自50个正常肝脏组织样本和373个HCC组织样本。通过差异基因表达分析发现7 230个在HCC和正常肝脏组织之间差异表达的基因(HCC中3 691个上调基因和3 539个下调基因)。富集分析表明,上调的差异表达基因主要参与细胞周期调控和有丝分裂过程;下调的差异表达基因主要参与小分子代谢和有机酸代谢等过程。WGCNA确定了19个与HCC患者临床特征相关基因模块,通过分析模块与临床特征之间的关系,筛选出青色模块和紫色模块。青色模块基因中同时与患者总生存期和无病生存期强烈相关的前两个基因为VPS45和FAM189B;紫色模块基因中同时与患者总生存期和无病生存期强烈相关的前两个基因分别为CLEC1B和FCN3,因此将VPS45、FAM189B、CLEC1B和FCN3确定为最终的Hub基因。人类蛋白质图谱数据库免疫组织化学染色显示:VPS45和FAM189B在HCC组织中的表达高于正常肝脏组织,FCN3在HCC组织中的表达低于正常肝脏组织,CLEC1B在HCC组织和正常肝脏组织中表达差异不明显。结论 初步确定VPS45、FAM189B、CLEC1B和FCN3可能是HCC的新型潜在生物标志物,这些Hub基因可能为HCC的靶向治疗提供理论基础。
【关键词】 癌,肝细胞;加权基因共表达网络分析;Hub基因;分子靶向治疗
【中图分类号】 R 730.261 【文献标识码】 A DOI:10.12114/j.issn.1007-9572.2023.0243
Analysis and Identification of Hub Genes in Hepatocellular Carcinoma Based on Weighted Gene Co-expression Network and Cancer Genome Atlas Clinical Data
CHEN Chao1,CHEN Tianxiang2,LIU Qianwei1,ZHANG Zhi1,WANG Huanhuan2,WU Pingping1,GAO Lei1,YU Zhaoxiang1*
1.Department of General Surgery,the First Affiliated Hospital of Xi'an Medical University,Xi'an 710077,China
2.Department of Hepatobiliary Surgery,the First Affiliated Hospital of Xi'an Jiaotong University,Xi'an 710061,China
*Corresponding author:YU Zhaoxiang,Professor/Chief physician;E-mail:yuzhaoxiang2@163.com
【Abstract】 Background Hepatocellular carcinoma(HCC) is the third leading cause of common cancer-related mortality globally,accounting for approximately 90% of all primary liver cancer cases. Its recurrence and mortality rates are high,with the underlying molecular mechanisms remaining unclear. Objective To explore potential molecular mechanisms of HCC and explore novel biomarkers. Methods RNA-seq expression data and clinical information were retrieved from TCGA database,differential gene expression analysis was conducted between normal liver tissue and HCC tissue. Enrichment analysis on the differentially expressed genes was performed. Based on the gene expression data profiles of HCC in TCGA,a co-expression network was established using the WGCNA R package,and weighted gene co-expression network analysis(WGCNA) was performed to select clinically significant modules and screen candidate Hub genes;the candidate Hub genes were further analyzed for significant differential expression in HCC tissues and normal liver tissues,and whether they were significantly correlated with the overall survival and disease-free survival of HCC patients. The Hub genes were conclusively identified,and their protein expression was validated through the Human Protein Atlas database. Results The genetic expression data in this study were obtained from 50 normal liver tissue samples and 373 HCC tissue samples. Through differential gene expression analysis,a total of 7 230 genes differential expression between HCC and normal hepatic tissue,comprising 3 691 up-regulated genes and 3 539 down-regulated genes in HCC were identified. Enrichment analysis showed that the up-regulated differentially expressed genes were mainly involved in cell cycle regulation and mitotic processes;the down-regulated differentially expressed genes were mainly involved in processes such as small molecule metabolism and organic acid metabolism. WGCNA identified 19 gene modules related to the clinical features of HCC patients,the cyan and purple modules were screened by analyzing the relationship between the modules and the clinical features. The first two genes in the cyan module genes that were strongly associated with both overall survival and disease-free survival of patients were VPS45 and FAM189B. In the purple module genes,first two genes that were strongly associated with both overall survival and disease-free survival of patients were CLEC1B and FCN3,respectively;therefore,VPS45,FAM189B,CLEC1B and FCN3 were identified as the final Hub genes. Immunohistochemical staining in the Human Protein Atlas database showed that VPS45 and FAM189B were expressed higher in HCC tissues than in normal liver tissues. FCN3 was expressed in HCC tissues lower than in normal liver tissues,the difference in the expression of CLEC1B between HCC tissues and normal liver tissues was not obvious. Conclusion VPS45,FAM189B,CLEC1B and FCN3 have been preliminary identified as possible novel potential biomarkers for HCC,which may provide a theoretical basis for targeted therapy of HCC.
【Key words】 Carcinoma,hepatocellular;Weighted gene co-expression network analysis;Hub gene;Molecular targeted therapy
肝细胞癌(HCC)是癌症相关死亡的第三大原因[1],也是中国近年来癌症相关死亡的主要原因。HCC在所有原发性肝癌病例中占90%[2]。HCC发病机制的主要危险因素包括慢性乙型肝炎病毒(HBV)和丙型肝炎病毒(HCV)感染、吸烟、饮酒、超重和非酒精性脂肪性肝病(NAFLD)、糖尿病以及黄曲霉毒素B1摄入量等[3],这些因素导致DNA损伤,表观遗传改变和癌症相关突变,最终导致HCC进展[4]。绝大多数患者确诊时已是晚期。尽管HCC的治疗在近几年取得很大进展,主要包括经导管动脉化疗栓塞、分子靶向治疗、消融治疗、手术切除和肝移植等[3,5],但HCC患者的5年生存率仍然很低[6],并且其5年复发率高达80%~90%,预后较差[7]。HCC发生和发展的确切机制仍不清楚,因此,阐明HCC发生、发展的分子机制,开发新的诊断和治疗方法来改善HCC的临床结果是非常迫切和必要的。
近年来,随着高通量测序技术不断发展,为癌症的基因组学、转录组学和表观基因组学的特征提供了新的研究方法。表达谱的生物信息学分析已被广泛用于鉴定新的和更有效的潜在生物标志物,用于癌症的治疗和患者预后[8]。通过生物信息学的大数据分析,可以有效整合复杂疾病,尤其是癌症的多个大规模数据集[9]。加权基因共表达网络分析(WGCNA)是一种先进的方法,其具有准确和高效的广泛基因分析优点,用于基于相似的基因表达模式构建共表达模块,并分析临床特征模块与不同基因组之间的相关性[10]。WGCNA已被广泛用于识别不同类型癌症相关的临床特征模块和Hub基因。如一项基于WGCNA研究将6个Hub基因和人肾细胞癌的进展以及患者的预后联系起来[11],鉴定了在侵袭性肾上腺皮质恶性肿瘤中高表达并与总生存期呈明显负相关的4个Hub基因(TOP2A、CHEK1、TTK和CENPA)[12]。
本研究通过差异基因表达分析和WGCNA对从TCGA和GEO数据库中HCC的mRNA数据在表达和功能水平上进行分析。然后进行功能富集分析,确定与HCC患者临床特征相关的模块,以了解这些共表达基因的潜在生物学功能。通过这些生物信息学分析鉴定出Hub基因,并结合生存分析、HCC组织和正常肝脏组织表达差异分析、免疫组织化学染色分析和文献分析进行验证。这些结果将有助于临床了解HCC的病因和潜在的分子机制,并为HCC提供新的治疗靶点或生物标志物。
1 资料与方法
1.1 数据来源和数据处理
从TCGA数据库[13](https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga)中下载HCC患者的RNA测序数据(50个正常样本和373个HCC样本)、临床信息和生存信息(59个正常样本和379个HCC样本),具体信息见附表1~3(扫描正文首页二维码查看),患者临床信息数据和样本量见表1。根据注释文档将探针转化为基因符号,通过测定所有对应探针的中位表达值,去除同一基因的重复探针。根据处理的结果,总共有11 627个基因被选中用于后续分析。分析的工作流程图见附录图1(扫描正文首页二维码查看)。
1.2 HCC中差异表达基因的筛选
使用R软件包UMAP(version 0.2.7.0)进行分析,对表达谱进行z-score,再使用UMAP函数进行降维分析以获得降维后的矩阵。Limma(linear models for microarray data)[14]是一种基于广义线性模型的差异表达筛选方法,用于识别正常肝脏组织和HCC组织之间的差异表达基因,在此处使用R软件包limma(version 3.40.6)进行差异分析,以获得正常肝脏组织与HCC组织的差异基因。获取的表达谱数据集,利用lmFit函数进行多元线性回归分析,进一步使用eBays函数进行分析,最终获得显著性差异的基因。
1.3 差异表达基因的功能富集
对于基因集进行功能富集分析,使用R软件包org.Hs.eg.db(version 3.1.0)中的基因GO注释,将其作为背景把基因映射到背景集合中,使用R软件包clusterProfiler(version 3.14.3)进行富集分析,从而获取基因集富集的结果。设定最小基因集为5,最大基因集为5 000,P<0.05和FDR<0.1作为显著性差异。
1.4 加权基因共表达网络分析
基于TCGA中HCC的基因表达数据概况,使用WGCNA R包建立共表达网络。以基因表达谱为例:先对所有成对基因进行Pearson相关矩阵和平均连锁法,然后使用幂函数amn=|Cmn|^β(Cmn=Gene_m和Gene_n)之间的Pearson's相关,构建出加权邻接矩阵。β是一个软阈值参数,其可以强调基因之间的强相关性。在选择5的幂之后,将邻接变换为拓扑重叠矩阵(topological overlap matrix,TOM)TOMi,j = (lij+ aij)/[min(ki+kj)+1-aij]。为了将具有相似表达谱的基因分类为基因模块,根据基于TOM的不相似性度量进行平均连锁分级聚类,基因树图的最小大小(基因组)为50。此外还合并了距离<0.25的模块,最终获得了19个共表达模块,其中grey模块被认为是无法被分配给任何模块的基因集合。
1.5 具有临床意义的模块的选择和HCC中的Hub基因的鉴定
基因表达和临床特征之间的线性关系被赋予一个基因显著性,等于单个基因P值的对数。如果基因显著性与模块成员密切相关,定义为模块的特征基因与个体基因表达谱之间的相关性,从而得出结论该模块的中心基因与HCC[15]相关,将这些基因视为候选Hub基因。如先前研究[16]所示:计算与基因的表达相关性以获得基因显著性,同时计算模块特征向量与基因的表达相关性以获得模块成员,根据截止标准,76个在临床显著模块中具有高连接性的基因被鉴定为候选Hub基因。
1.6 Hub基因的基因验证和生物信息学验证
使用GEPIA在线网站(http://gepia.cancer-pku.cn/)分析HCC样本中Hub基因的表达水平,并基于Kaplan-Meier分析使用R套件中的生存包(版本:3.2-7)。从TCGA获得359个HCC肿瘤样本的差异基因表达谱和预后数据,然后确定每个基因的中位数表达值,根据给定基因的表达水平是高于还是低于中位数,样本被分配到给定基因的“高表达”或“低表达”组。使用对数秩检验评估高表达组或低表达组之间的总体生存期和无病生存期的显著性。如果P<0.05,认为该基因是经过验证的Hub基因。根据来自TCGA和GEPIA网站上的数据,筛选正常肝脏组织和HCC组织之间Hub基因表达的差异。以P<0.05为差异有统计学意义。
1.7 人类蛋白质图谱数据库对Hub基因蛋白表达的验证
人类蛋白质图谱数据库(https://www.proteinatlas.org/)主要用于为提供各种人类蛋白质的组织和细胞分布信息。使用来自人类蛋白质图谱数据库的免疫组织化学染色结果验证了HCC组织和正常肝脏组织之间生存相关基因的蛋白质表达。
2 结果
2.1 数据预处理
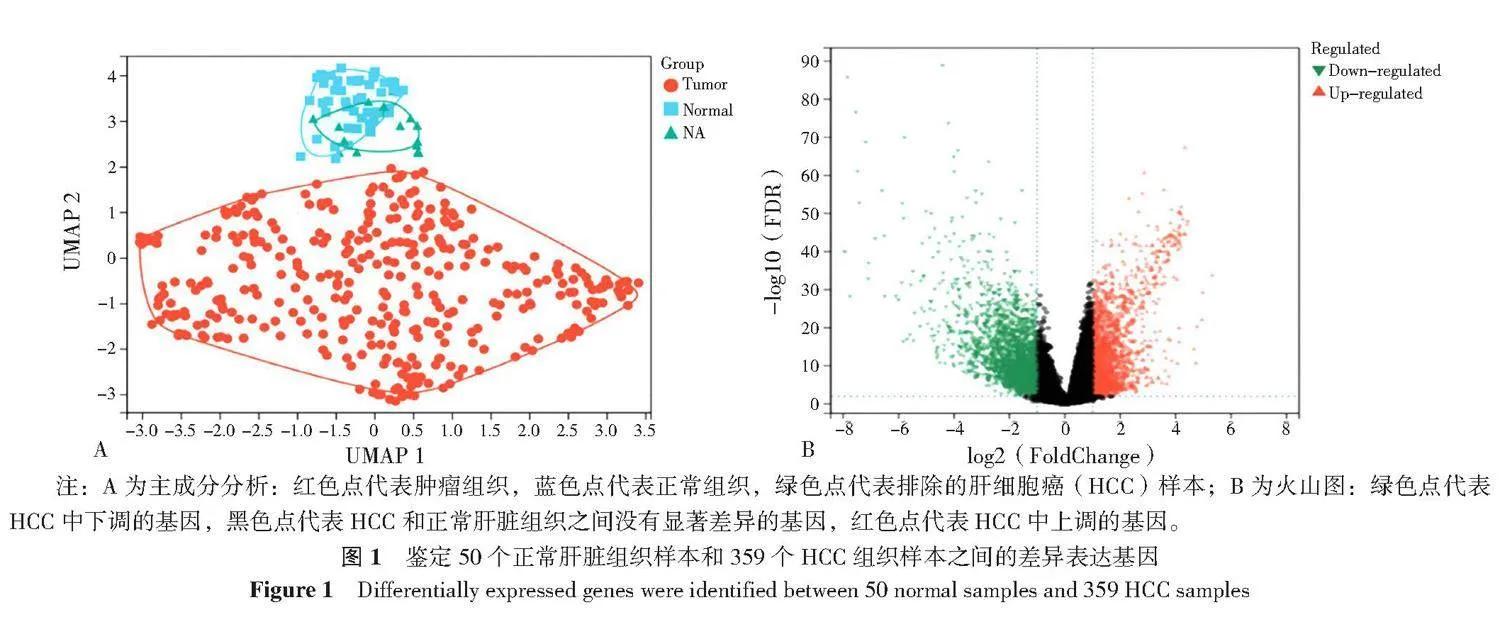
本研究的表达数据来自50个正常肝脏组织样本和373个HCC组织样本(附表4)。基于主成分分析从数据中排除了14个肿瘤样本(图1A)。最终409个样本的基因表达谱用于后续分析。
2.2 HCC样本中差异表达基因的鉴定及GO富集分析
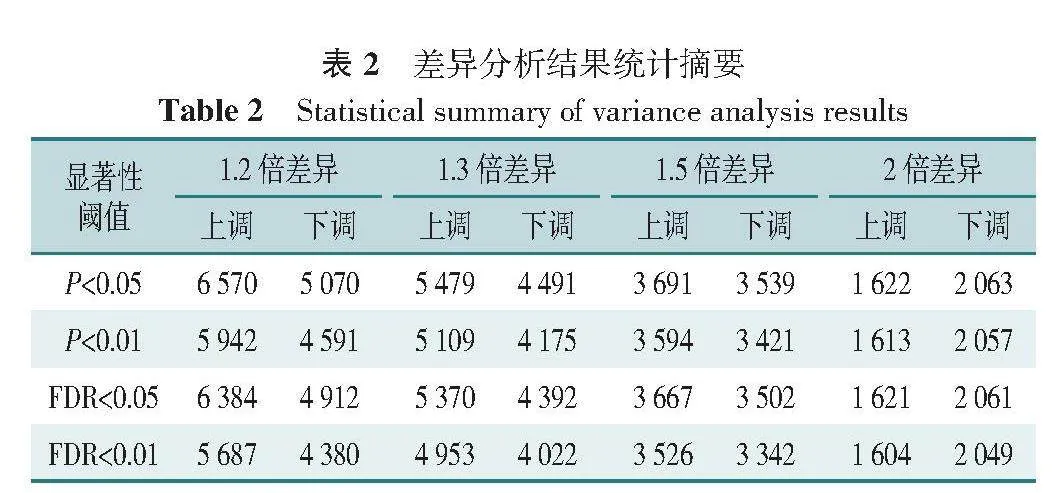
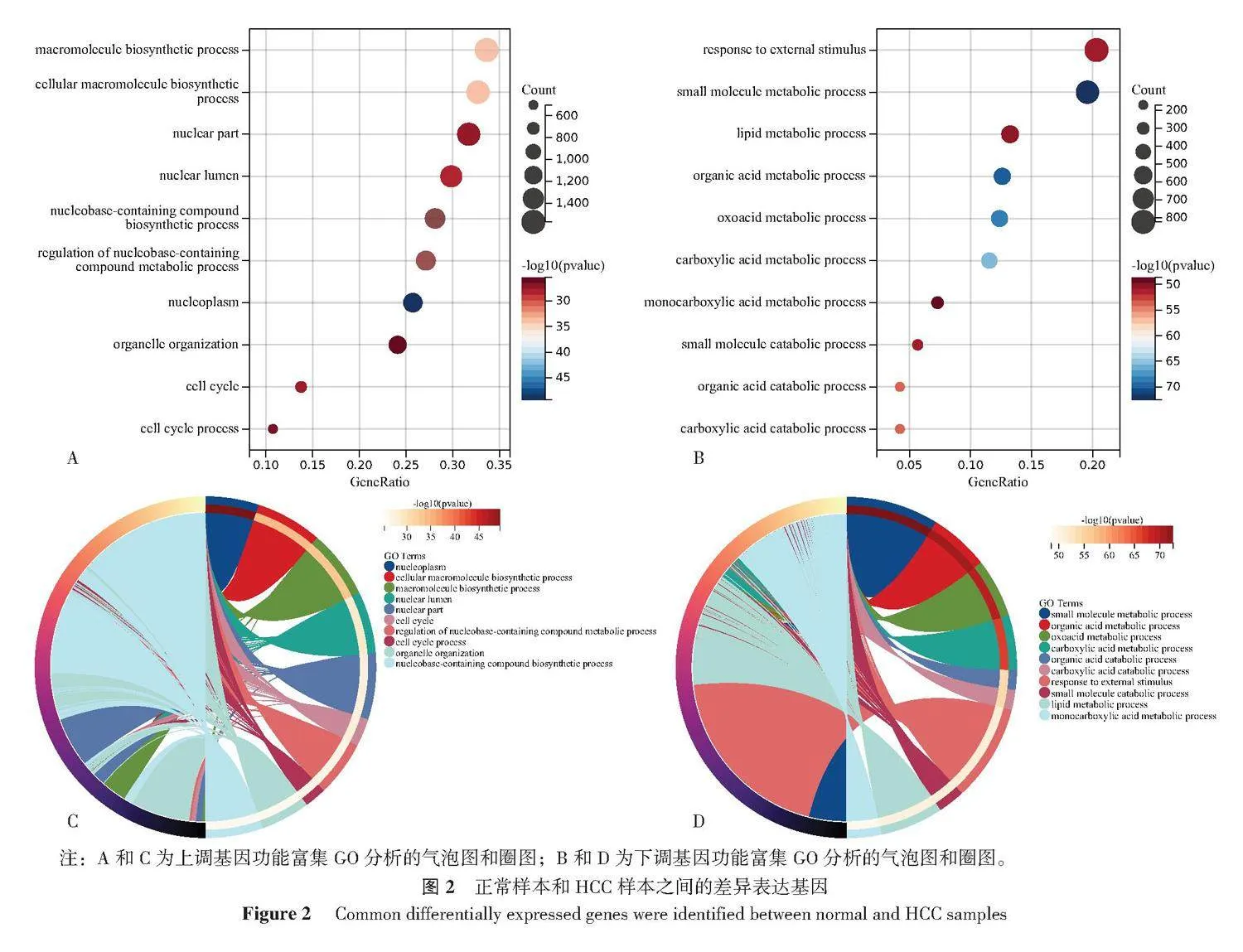
在50个正常样本和359个HCC样本之间共鉴定出7 230个差异表达基因,差异表达基因火山图见图1B,包括3 691个上调基因和3 539个下调基因(附表5),差异分析结果见表2。为了探索差异表达基因在HCC中的潜在生物学功能,本研究对其进行GO富集分析,结果见附表6和附表7。
上调的差异表达基因主要参与细胞周期调控、有丝分裂过程、核分裂和染色体分离等(图2A、2C);下调的差异表达基因主要参与对外刺激反应、小分子代谢和有机酸代谢等过程(图2B、2D)。
比较HCC组织和正常肝脏组织的基因表达差异,获得表达数据集,根据差异倍数为1.5倍,P<0.05为显著差异,进一步利用lmFit函数进行多元线性回归分析,再使用eBays函数进行分析,最终获得每个基因的差异显著性,并绘制差异表达基因热图(附录图2)。
2.3 WGCNA和关键模块的识别
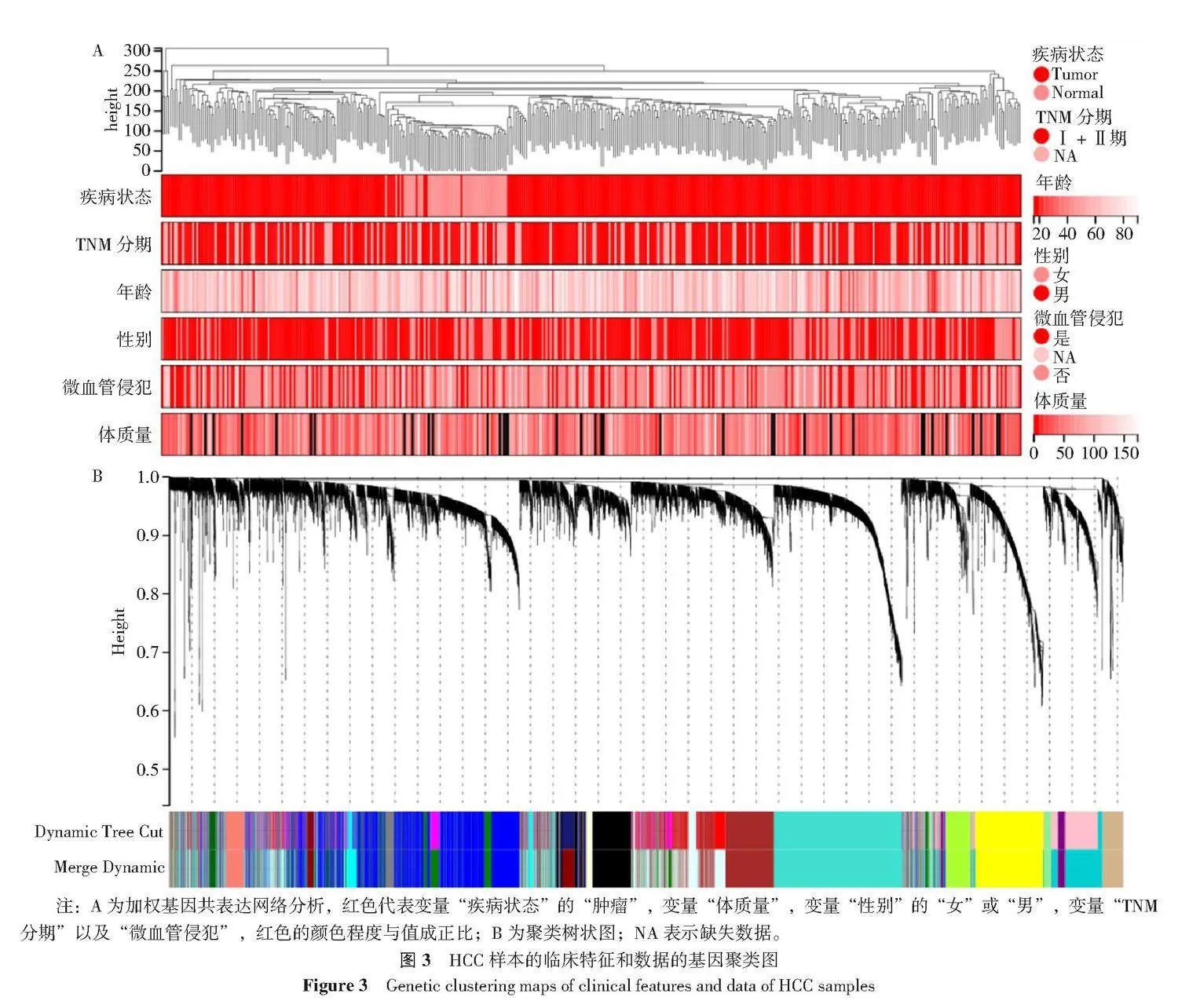
加权基因共表达网络分析基于7 230个差异表达基因的表达矩阵和409个HCC样本的临床数据。进行聚类分析检查409个样本的数据质量,结果显示所有样本在聚类中并且在截止阈值内,加权基因共表达网络分析中应用了6个临床变量:疾病状态、性别、年龄、体质量、TNM分期和微血管侵犯(图3A)。409个样本分为两个簇,肿瘤和正常,HCC样本的临床特征和数据的聚类树状图(图3B)。
为了构建无标度网络,将软阈值β设置为5,独立度设置为0.86,平均连通性接近0(图4A、4B)。模块参数设置为最小模块30,敏感性为3,模块合并阈值为0.25,将其中具有相似表达模式的差异表达基因聚集到相同的模块中,此外合并距离<0.25的模块,最终获得了19个共表达模块(附录图3),并绘制模块特征基因热图(图4C),其中grey模块是无法被分配给任何模块的基因集合,差异基因表达谱及差异基因见附表8。
然后尝试评估模块与临床特征之间的关系,发现青色模块的特征基因与HCC之间具有强正相关(cor=0.64,P=3.6×10-50),而紫色模块的特征基因与HCC之间具有高度负相关(cor=-0.80,P=5.6×10-97)(附录图4)。结果表明,青色模块可能促进HCC的肿瘤发生,而紫色模块可能会预防HCC。因此,分析青色模块和紫色模块的Hub基因。
2.4 从青色和紫色模块中识别候选Hub基因
MM和GS分数在青色和紫色模块中彼此呈正相关(图5)。青色模块中选择Hub基因的标准相对低于标准截止阈值(MM>0.8)。在青色模块中,选择满足“cor.gene Module Membership”>0.7和“cor.gene Trait Significance”>0.5阈值的前8个基因,分别为TOMM40L、VPS45、MSTO1、FAM189B、TTC13、PYGO2、NVL和EHMT2。在紫色模块中,选择满足“cor.gene Module Membership”>0.8和“cor.gene Trait Significance”>0.7阈值的前16个基因,分别为CLEC4M、BMP10、CLEC1B、CLEC4G、GDF2、NDST3、BMPER、STAB2、CCL23、CHRM2、COL6A6、CRHBP、FCN3和CCBE1。
2.5 Hub基因表达及其与生存的相关性
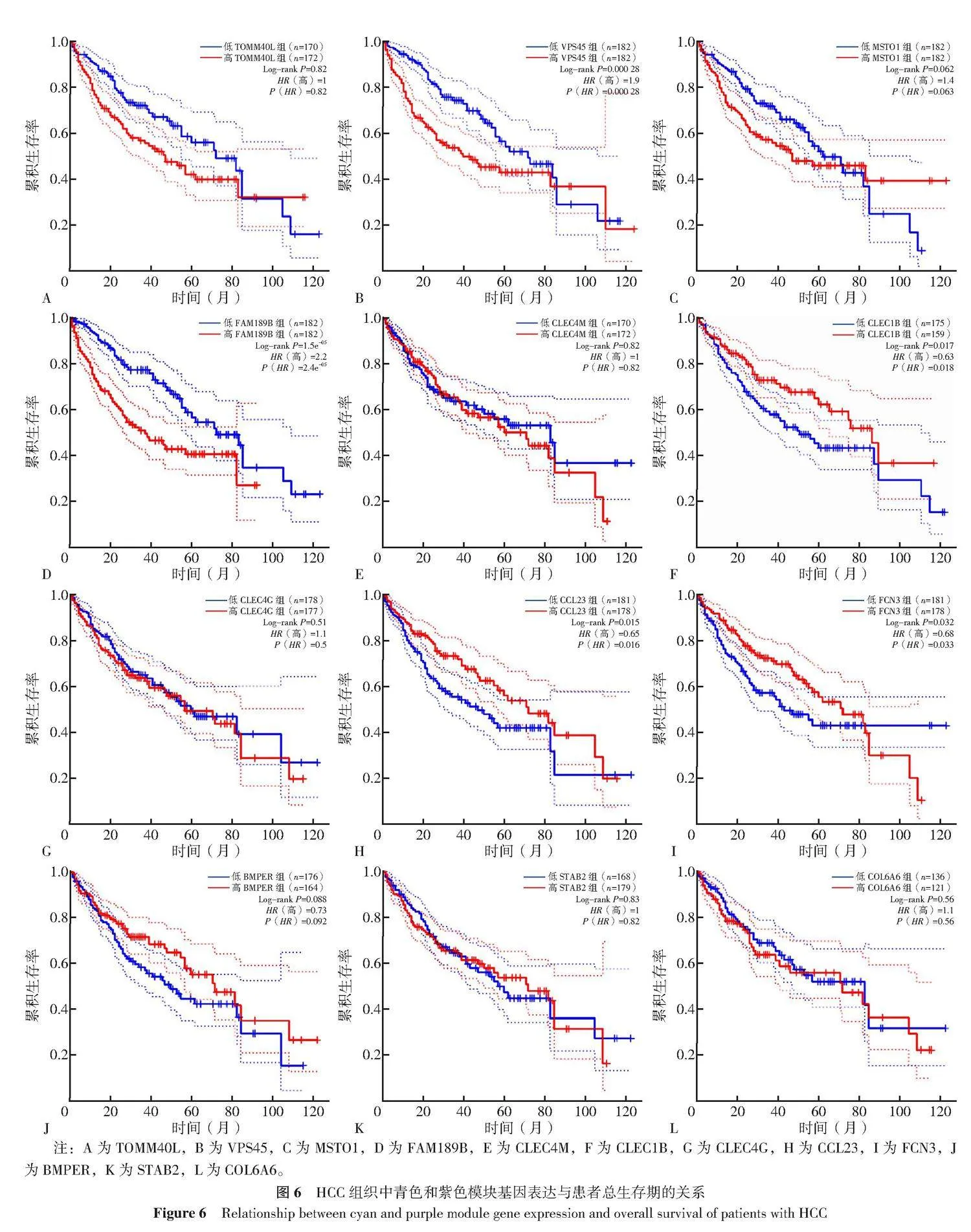
青色模块基因中仅与患者总生存期强相关的前3个基因分别为TOMM40L、VPS45和FAM189B;青色模块基因中同时与患者总生存期和无病生存期强相关的前两个基因为VPS45和FAM189B。紫色模块基因中仅与患者总生存期强相关的前3个基因分别为CLEC1B、CCL23和FCN3;紫色模块基因中同时与患者总生存期和无病生存期强相关的前两个基因分别为CLEC1B和FCN3(图6)。因此,将VPS45、FAM189B、CLEC1B和FCN3确定为最终的Hub基因。两个模块中与无病生存期有显著差异的基因有VPS45、FAM189B、CLEC1B、FCN3和BMPER(附录图5)。
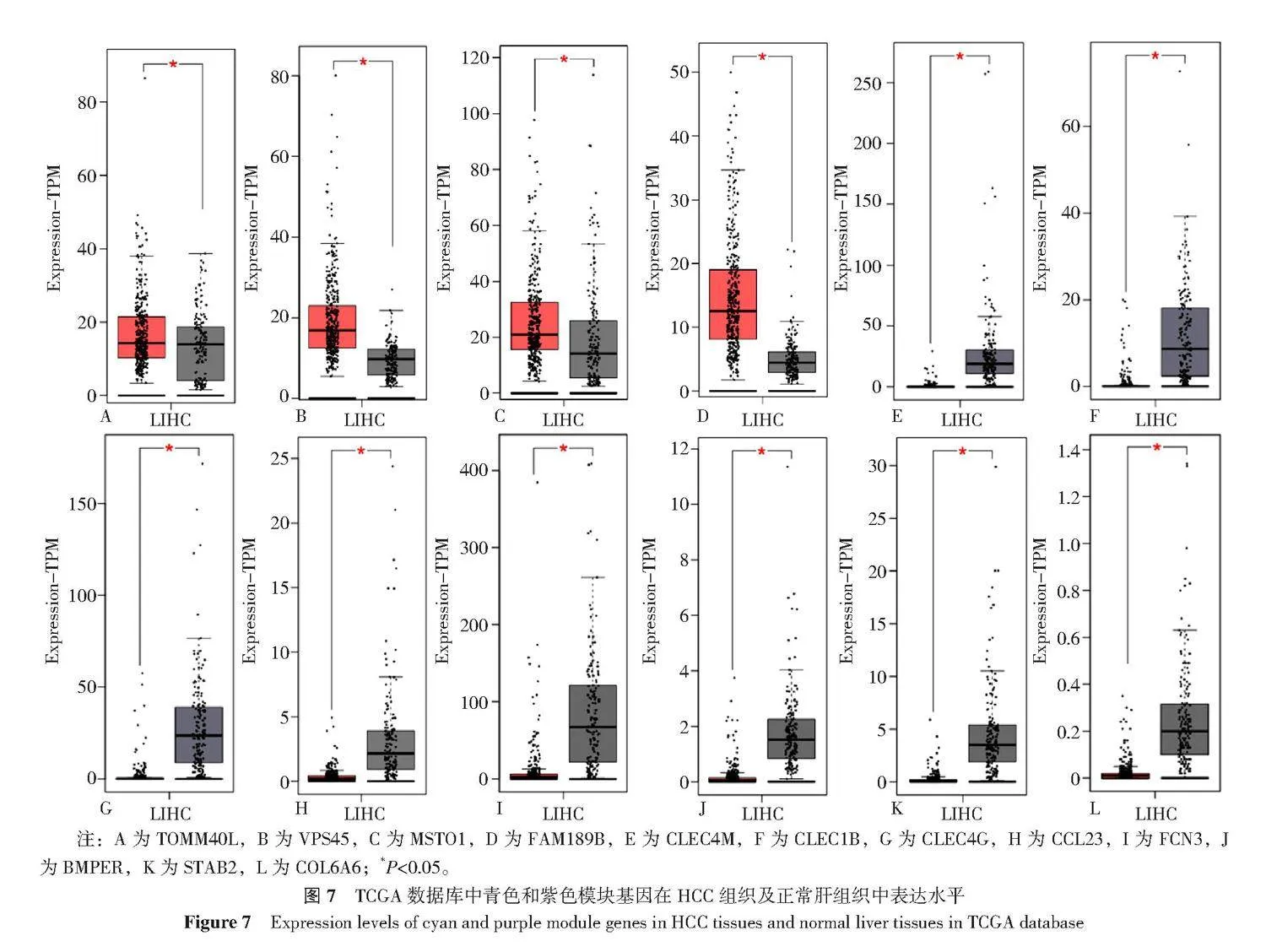
使用GEPIA网站,分析这些模块基因在HCC组织和正常组织之间表达情况(图7),结果显示,VPS45和FAM189B在HCC组织中上调,而CLEC1B和FCN3在HCC组织中下调。使用GEO数据库的数据获得了类似的结果(附录图6)。
2.6 免疫组织化学染色验证
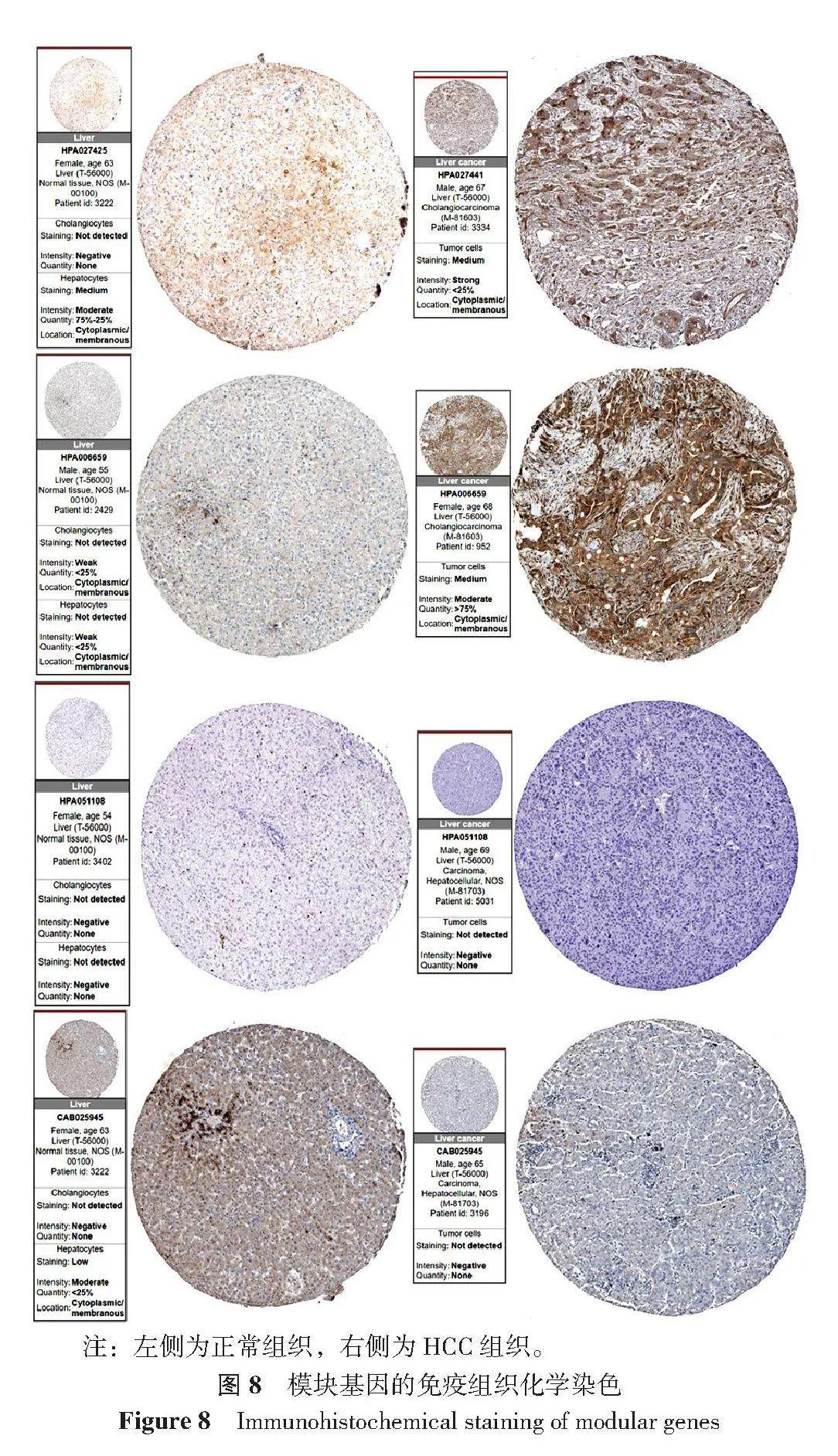
从人类蛋白质图谱(The Human Protein Atlas,HPA)数据库[17](https://www.proteinatlas.org)中获得的免疫组织化学染色显示:VPS45和FAM189B在HCC组织中的表达高于正常肝脏组织,而FCN3在HCC组织中的表达低于正常肝脏组织,CLEC1B在HCC组织和正常肝脏组织中表达差异不明显(图8)。
3 讨论
HCC筛查指对符合HCC高风险的患者定期进行检查,其目标是在早期时间检测出HCC并予以及时干预,使患者存活率和生存质量提高。在HCC的临床应用中,常见的传统筛查方式包括肝脏超声检查和血清甲胎蛋白,其灵敏度和特异度相对有限。因此,开发出新的更准确的检测方式十分具有临床应用价值。目前研究表明几个基因已被确定为HCC诊断的新型生物标志物。例如,长链非编码RNA KDM4A-AS1在许多肿瘤中过度表达,尤其是在HCC中,长链非编码RNA KDM4A-AS1水平与疾病分期和肿瘤分级呈正相关,与患者生存呈负相关[18],LV等[19]研究显示BAI3和CKAP2L可能是结直肠癌的潜在预后因子和治疗靶点。
在这项研究中,将差异表达基因和加权基因共表达网络结合起来,以提高识别HCC相关基因的能力。差异基因功能富集显示,主要参与细胞周期调控的基因在HCC组织中失调,与近些年相关研究结果一致[20]。正常细胞周期的紊乱是导致癌症的原因之一,靶向调节癌细胞周期是一种潜在的治疗方法[21]。生物信息学分析基于TCGA和GEO数据库,分析在HCC中重要模块和Hub基因,确定了两个关键模块,分别为青色模块和紫色模块。分析这两个模块基因与患者总生存期和无病生存期的相关性,选择了青色模块中显著相关的前两个基因VPS45和FAM189B,选择了紫色模块中显著相关的前两个基因CLEC1B和FCN3,这4个基因作为入选的Hub基因。
VPS45缺陷型中性粒细胞和成纤维细胞表面的β1整合素水平降低,VPS45缺陷型成纤维细胞运动能力受损和细胞凋亡增加[22]。另外有研究显示VPS45的表达水平和多种癌症相关,比如YAMANOI等[23]研究表明VPS45表达水平和恶性肿瘤卵巢癌相关。FAM189B又称为COTE1,通过机制分析表明,FAM189B可以与肿瘤抑制因子结构域氧化还原酶发生物理关联,FAM189B与HCC细胞的侵袭密切相关[24];FAM189B蛋白和mRNA的过表达可能会增加胃癌的发生率,并且细胞周期的通路是其KEGG富集分析最具有显著差异的通路[25]。CLEC1B是C型凝集素结构域家族1成员,并且其与肝细胞癌的免疫浸润相关,过表达的CLEC1B抑制HuH7细胞的增殖和迁移能力[26]。FCN3是一种分泌型凝集素,能够激活补体通路,FCN3的异位表达能够激活内质网应激未折叠蛋白,抑制内质网应激反应可提高肺腺癌细胞的存活率,FCN通过诱导内质网应激发挥抑癌作用[27]。本研究方法具有数据挖掘的局限性,研究所用数据来源于网络数据库,数据样本可能存在偏倚,测序结果在技术上可能存在偏差,为了提高WGCNA结果的可靠性,使用人类蛋白质图谱的免疫组织化学数据进行确认,然而由于数据库的限制,无法获得每个基因的肿瘤和邻近正常样本的所有相关IHC数据。获得的Hub基因在肝癌预后中的分子调节机制还需要进一步通过临床数据和基础实验来证实,其可能作为肝癌早期筛查分子标志物、预后分子标志物及肝癌治疗靶点,为人群筛查或肝癌患者治疗提供帮助。
总之,本研究进行了全面的生物信息学分析,以确定HCC和正常肝脏组织之间的潜在生物预测标志物,本研究结果表明,VPS45、FAM189B、CLEC1B和FCN3可能是HCC的新型潜在生物标志物,具有特殊临床意义,然而需要在大量临床样本中进行验证。
作者贡献:陈超提出研究目标,负责数据分析和写作;陈天翔负责数据验证;刘钱伟、张秩、王欢欢、吴平平和高磊负责数据整理;于照祥负责文章的质量控制与审查,对文章整体负责,监督管理。
本文无利益冲突。
参考文献
SUNG H,FERLAY J,SIEGEL R L,et al. Global cancer statistics 2020:GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin,2021,
71(3):209-249. DOI:10.3322/caac.21660.
MEHTA A,HERRERA H,BLOCK T. Glycosylation and liver cancer[J]. Adv Cancer Res,2015,126:257-279. DOI:10.1016/bs.acr.2014.11.005.
VOGEL A,MEYER T,SAPISOCHIN G,et al. Hepatocellular carcinoma[J]. Lancet,2022,400(10360):1345-1362. DOI:10.1016/s0140-6736(22)01200-4.
Cancer Genome Atlas Research Network Electronic Address. Comprehensive and integrative genomic characterization of hepatocellular carcinoma[J]. Cell,2017,169(7):1327-1341.e23. DOI:10.1016/j.cell.2017.05.046.
LLOVET J M,REAL M I,MONTAÑA X,et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma:a randomised controlled trial[J]. Lancet,2002,359(9319):1734-1739. DOI:10.1016/S0140-6736(02)08649-X.
SIEGEL R L,MILLER K D,JEMAL A. Cancer statistics,2017[J]. CA Cancer J Clin,2017,67(1):7-30.
XIA F,WU L L,LAU W Y,et al. Adjuvant sorafenib after heptectomy for Barcelona clinic liver cancer-stage C hepatocellular carcinoma patients[J]. World J Gastroenterol,2016,22(23):5384-5392. DOI:10.3748/wjg.v22.i23.5384.
LI N,ZHAN X Q. Identification of clinical trait-related lncRNA and mRNA biomarkers with weighted gene co-expression network analysis as useful tool for personalized medicine in ovarian cancer[J]. EPMA J,2019,10(3):273-290. DOI:10.1007/s13167-019-00175-0.
KUENZI B M,IDEKER T. A census of pathway maps in cancer systems biology[J]. Nat Rev Cancer,2020,20(4):233-246.
LANGFELDER P,HORVATH S. WGCNA:an R package for weighted correlation network analysis[J]. BMC Bioinformatics,2008,9:559. DOI:10.1186/1471-2105-9-559.
YUAN L S,CHEN L,QIAN K Y,et al. Co-expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC)[J]. Genom Data,2017,14:132-140. DOI:10.1016/j.gdata.
2017.10.006.
XIA W X,YU Q,LI G H,et al. Identification of four hub genes associated with adrenocortical carcinoma progression by WGCNA[J]. PeerJ,2019,7:e6555.
COLAPRICO A,SILVA T C,OLSEN C,et al. TCGAbiolinks:an R/Bioconductor package for integrative analysis of TCGA data[J]. Nucleic Acids Res,2016,44(8):e71.
RITCHIE M E,PHIPSON B,WU D,et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies[J]. Nucleic Acids Res,2015,43(7):e47.
DI Y,CHEN D S,YU W,et al. Bladder cancer stage-associated hub genes revealed by WGCNA co-expression network analysis[J]. Hereditas,2019,156:7. DOI:10.1186/s41065-019-0083-y.
TANG J N,KONG D G,CUI Q X,et al. Prognostic genes of breast cancer identified by gene co-expression network analysis[J]. Front Oncol,2018,8:374. DOI:10.3389/fonc.2018.00374.
KARLSSON M,ZHANG C,MÉAR L,et al. A single-cell type transcriptomics map of human tissues[J]. Sci Adv,2021,7(31):eabh2169. DOI:10.1126/sciadv.abh2169.
CHEN T X,LIU R K,NIU Y S,et al. HIF-1α-activated long non-coding RNA KDM4A-AS1 promotes hepatocellular carcinoma progression via the miR-411-5p/KPNA2/AKT pathway[J]. Cell Death Dis,2021,12(12):1152.
LV Y M,XIE B B,BAI B J,et al. Weighted gene coexpression analysis indicates that PLAGL2 and POFUT1 are related to the differential features of proximal and distal colorectal cancer[J]. Oncol Rep,2019,42(6):2473-2485.
SCHAFER K. The cell cycle:a review[J]. Vet Pathol,1998,35:461-478. DOI:10.1177/030098589803500601.
AARTS M,LINARDOPOULOS S,TURNER N C. Tumour selective targeting of cell cycle kinases for cancer treatment[J]. Curr Opin Pharmacol,2013,13(4):529-535. DOI:10.1016/j.coph.2013.03.012.
VILBOUX T,LEV A,MALICDAN M C V,et al. A congenital neutrophil defect syndrome associated with mutations in VPS45[J]. N Engl J Med,2013,369(1):54-65.
YAMANOI K,BABA T,ABIKO K,et al. Acquisition of a side population fraction augments malignant phenotype in ovarian cancer[J]. Sci Rep,2019,9(1):14215. DOI:10.1038/s41598-019-50794-w.
ZHANG H,HUANG C J,TIAN Y,et al. Ectopic overexpression of COTE1 promotes cellular invasion of hepatocellular carcinoma[J]. Asian Pac J Cancer Prev,2012,13(11):5799-5804. DOI:10.7314/apjcp.2012.13.11.5799.
WU C L,TAN Q X,LIU D,et al. High FAM189B expression and its prognostic value in patients with gastric cancer[J]. Biomed Res Int,2021,2021:8875971. DOI:10.1155/2021/8875971.
ZHANG G H,SU L S,LV X P,et al. A novel tumor doubling time-related immune gene signature for prognosis prediction in hepatocellular carcinoma[J]. Cancer Cell Int,2021,21(1):522. DOI:10.1186/s12935-021-02227-w.
JANG H,JUN Y,KIM S,et al. FCN3 functions as a tumor suppressor of lung adenocarcinoma through induction of endoplasmic reticulum stress[J]. Cell Death Dis,2021,12(4):407.
(收稿日期:2023-03-17;修回日期:2023-08-18)
(本文编辑:贾萌萌)
基金项目:陕西省科技厅面上项目(2021JM-503)
引用本文:陈超,陈天翔,刘钱伟,等. 基于加权基因共表达网络和癌症基因组图谱临床数据分析并鉴定肝细胞癌的Hub基因研究[J]. 中国全科医学,2024,27(32):4050-4059. DOI:10.12114/j.issn.1007-9572.2023.0243. [www.chinagp.net]
CHEN C,CHEN T X,LIU Q W,et al. Analysis and identification of Hub genes in hepatocellular carcinoma based on weighted gene co-expression network and cancer genome atlas clinical data[J]. Chinese General Practice,2024,27(32):4050-4059.
© Editorial Office of Chinese General Practice. This is an open access article under the CC BY-NC-ND 4.0 license.
